NEULASTATM
pegfilgrastim
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p seringue | |
| Pegfilgrastim* | 6 mg |
La concentration exprimée en protéine pure est de 10 mg/ml. La concentration est de 20 mg/ml lorsque la fraction pégylée (PEG) est prise en compte.
L'activité de ce médicament ne doit pas être comparée à celle d'autres protéines pégylées ou non pégylées de la même classe thérapeutique. Pour plus d'informations, cf Pharmacodynamie.
* Produit sur des cellules d'Escherichia coli par la technique de l'ADN recombinant suivie d'une conjugaison au polyéthylèneglycol (PEG).
** Excipients ayant un effet notoire (cf Mises en garde et Précautions d'emploi).
INDICATIONS |
POSOLOGIE ET MODE D'ADMINISTRATION |
Une dose de 6 mg (en une seringue unique préremplie) de Neulasta est recommandée pour chaque cycle de chimiothérapie, en administration sous-cutanée environ 24 heures après la fin de la chimiothérapie cytotoxique.
- Enfant :
- Les données concernant l'administration chez les enfants sont limitées. Cf Effets indésirables, Pharmacodynamie, Pharmacocinétique.
- Insuffisance rénale :
- Aucune adaptation posologique n'est recommandée chez les patients insuffisants rénaux, y compris ceux présentant une maladie rénale à un stade avancé.
CONTRE-INDICATIONS |
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
Le facteur de croissance de la lignée granulocytaire peut stimuler la croissance des cellules myéloïdes in vitro et des effets similaires ont pu être observés sur certaines cellules non myéloïdes in vitro.
La tolérance et l'efficacité de Neulasta n'ont pas été étudiées chez les patients atteints de syndrome myélodysplasique, de leucémie myéloïde chronique ou de leucémie aiguë myéloïde (LAM) secondaire ; par conséquent, Neulasta ne doit pas être utilisé chez ces patients. Il importe de bien différencier le diagnostic d'une transformation blastique d'une leucémie myéloïde chronique de celui d'une leucémie aiguë myéloïde.
La tolérance et l'efficacité de Neulasta n'ont pas été établies chez les patients de moins de 55 ans atteints de LAM de novo et présentant une cytogénétique t(15;17).
La tolérance et l'efficacité de Neulasta n'ont pas été étudiées chez les patients recevant une chimiothérapie à haute dose.
Après administration de facteurs de croissance de la lignée granulocytaire (G-CSFs), de rares cas d'effets indésirables pulmonaires ont été rapportés (>= 1/10 000 à < 1/1000), en particulier, des pneumonies interstitielles. Les risques peuvent être majorés chez les patients ayant des antécédents récents d'infiltration pulmonaire ou de pneumonie.
L'apparition de signes pulmonaires, tels que toux, fièvre et dyspnée, associés à des signes radiologiques d'infiltration pulmonaire avec détérioration de la fonction respiratoire et augmentation du nombre de polynucléaires neutrophiles peuvent être des signes préliminaires d'un syndrome de détresse respiratoire de l'adulte (ARDS). Dans de telles circonstances, Neulasta doit être arrêté après avis du médecin et un traitement approprié doit être institué.
Après administration de pegfilgrastim, des cas fréquents (>= 1/100 à < 1/10) et généralement asymptomatiques de splénomégalie, ainsi que de très rares cas (< 1/10 000) de rupture splénique pouvant entraîner une issue fatale, ont été observés. Par conséquent, le volume de la rate doit être surveillé attentivement (par exemple examen clinique, échographie). Un diagnostic de rupture splénique devra être envisagé chez des patients présentant une douleur au niveau de l'hypochondre gauche ou une douleur au sommet de l'épaule.
Un traitement par Neulasta seul ne prévient pas la thrombopénie et l'anémie dues au maintien d'une chimiothérapie myélosuppressive aux doses et délais prévus. Une surveillance régulière du nombre de plaquettes et de l'hématocrite est recommandée.
Neulasta ne doit pas être utilisé pour augmenter les doses de chimiothérapie cytotoxique au-delà des schémas posologiques établis.
Des crises drépanocytaires ont été associées à une utilisation de pegfilgrastim chez des patients atteints d'anémie falciforme. Par conséquent, Neulasta doit être administré avec précaution chez les patients atteints d'anémie falciforme et une surveillance étroite des paramètres cliniques et biologiques doit être instituée. Il faut être attentif au lien éventuel entre Neulasta et la survenue d'une splénomégalie ou d'un accident veino-occlusif.
Un nombre de leucocytes égal ou supérieur à 100 × 109/l a été observé chez moins de 1 % des sujets recevant Neulasta. Aucun effet indésirable directement attribuable à ce niveau de leucocytose n'a été rapporté. Une telle élévation de leucocytes est transitoire, spécifiquement observée 24 à 48 heures après l'administration et conforme aux effets pharmacodynamiques de Neulasta.
La tolérance et l'efficacité de Neulasta pour la mobilisation de cellules souches progénitrices (CSP) dans le sang circulant chez des patients ou des donneurs sains n'ont pas été suffisamment évaluées.
Le capuchon de la seringue préremplie contient du caoutchouc naturel sec (un dérivé du latex) pouvant entraîner des réactions allergiques.
L'augmentation de l'activité hématopoïétique de la moelle osseuse en réponse à un traitement par facteurs de croissance a été associée à des variations transitoires observables de la scintigraphie osseuse. Celles-ci doivent être prises en compte lors de l'interprétation des résultats de la scintigraphie osseuse.
En raison de la présence de sorbitol, Neulasta ne doit pas être utilisé chez les patients présentant des troubles héréditaires rares d'intolérance au fructose.
Neulasta contient moins de 1 mmol (23 mg) de sodium par dose de 6 mg. Il est donc pratiquement exempt de sodium.
INTERACTIONS |
Les interactions éventuelles avec d'autres facteurs de croissance hématopoïétiques et avec les cytokines n'ont pas été spécifiquement étudiées au cours des essais cliniques.
L'interaction potentielle avec le lithium, qui favorise également la libération des neutrophiles, n'a pas été spécifiquement étudiée. Aucun élément ne permet d'affirmer l'existence d'un effet indésirable dû à cette interaction.
La tolérance et l'efficacité de Neulasta n'ont pas été évaluées chez les patients recevant une chimiothérapie entraînant une myélosuppression retardée, par exemple les nitrosourées.
Des études spécifiques d'interactions médicamenteuses ou de métabolisme n'ont pas été réalisées. Cependant, les études cliniques n'ont pas mis en évidence d'interaction entre Neulasta et d'autres médicaments.
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Neulasta ne doit pas être utilisé pendant la grossesse sauf en cas de nécessité absolue.
En l'absence de données cliniques chez la femme en période d'allaitement, il est recommandé de ne pas administrer Neulasta chez la femme qui allaite.
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
L'effet indésirable très fréquent, le plus souvent rapporté, lié à l'administration du produit à l'étude, a été la douleur osseuse. La douleur osseuse a été en général d'intensité légère à modérée, transitoire, et a pu être contrôlée chez la plupart des patients par l'administration d'antalgiques classiques.
Des réactions de type allergique, incluant anaphylaxie, rash cutané, urticaire, angio-oedème, dyspnée, hypotension, réactions au site d'injection, érythème et bouffées vasomotrices apparaissant au cours de l'administration initiale ou de la poursuite du traitement, ont été rapportées avec Neulasta. Dans certains cas, la réadministration du produit a entraîné la réapparition des symptômes, suggérant ainsi une relation de cause à effet. Si une réaction allergique grave survient, un traitement approprié devra être administré, associé à un suivi étroit du patient pendant plusieurs jours. Le pegfilgrastim doit être définitivement arrêté chez les patients présentant une réaction allergique grave.
Des augmentations réversibles, légères à modérées, des concentrations d'acide urique et de phosphatases alcalines, sans signes cliniques associés, ont été fréquentes (>= 1/100 à < 1/10) ; des augmentations réversibles, légères à modérées, de la concentration de lactate-déshydrogénase, sans signes cliniques associés, ont été très fréquentes (>= 1/10) chez des patients recevant Neulasta à la suite d'une chimiothérapie cytotoxique. Des nausées ont été observées chez des volontaires sains et chez des patients recevant une chimiothérapie.
Après administration de pegfilgrastim, des cas fréquents (>= 1/100 à < 1/10) et généralement asymptomatiques d'augmentation du volume de la rate, ainsi que de très rares cas de rupture splénique pouvant entraîner une issue fatale, ont été observés (cf Mises en garde et Précautions d'emploi). Les autres effets indésirables fréquemment rapportés ont été : douleurs, douleur au site d'injection, douleur thoracique (non cardiaque), céphalées, arthralgie, myalgie, rachialgie, douleur des membres, douleur osseuse et cervicalgie.
De rares cas (>= 1/10 000 à < 1/1000) d'effets indésirables pulmonaires incluant pneumonie interstitielle, oedème pulmonaire, infiltration et fibrose pulmonaires, ont été rapportés. Certains de ces cas ont entraîné une insuffisance respiratoire ou un syndrome de détresse respiratoire de l'adulte (ARDS) pouvant entraîner une issue fatale (cf Mises en garde et Précautions d'emploi).
De rares cas (>= 1/10 000 à < 1/1000) de thrombopénie et d'hyperleucocytose ont été rapportés.
De rares cas (>= 1/10 000 à < 1/1000) de syndrome de Sweet ont été rapportés, bien que certains cas puissent être reliés à la pathologie maligne hématologique sous-jacente.
De très rares cas (< 1/10 000) de vascularites cutanées ont été rapportés chez les patients traités par Neulasta. Le mécanisme de la vascularite chez les patients recevant Neulasta n'est pas connu.
De très rares cas (< 1/10 000) d'anomalies des tests de la fonction hépatique : augmentation des ALAT (alanine aminotransférase) ou des ASAT (aspartate aminotransférase), ont été observés chez les patients ayant reçu du pegfilgrastim après chimiothérapie cytotoxique. Ces augmentations étaient transitoires et les valeurs sont revenues à la normale.
Des cas isolés de crises drépanocytaires ont été rapportés chez des patients atteints d'anémie falciforme (cf Mises en garde et Précautions d'emploi).
- Enfant :
- Il a été observé une fréquence plus élevée d'événements indésirables graves chez les jeunes enfants de 0 à 5 ans (92 %) comparé aux enfants âgés de 6 à 11 ans et de 12 à 21 ans respectivement (80 % et 67 %) et aux adultes. L'effet indésirable le plus fréquent était la douleur osseuse (cf Pharmacodynamie, Pharmacocinétique).
SURDOSAGE |
PHARMACODYNAMIE |
Classe pharmacothérapeutique : cytokines (code ATC : L03AA13).
Le Granulocyte-Colony Stimulating Factor humain (facteur de croissance de la lignée granulocytaire, G-CSF) est une glycoprotéine qui régule la production et la libération des polynucléaires neutrophiles à partir de la moelle osseuse.
Le pegfilgrastim est une forme conjuguée covalente de G-CSF humain recombinant (r-metHuG-CSF) attaché à une molécule de polyéthylèneglycol (PEG) de 20 kd. Le pegfilgrastim est une forme à durée prolongée de filgrastim, par diminution de la clairance rénale. Le pegfilgrastim et le filgrastim présentent un mécanisme d'action identique, entraînant une augmentation marquée, dans les 24 heures, du nombre de polynucléaires neutrophiles circulants, ainsi qu'une augmentation mineure des monocytes et/ou des lymphocytes. Comme pour le filgrastim, les neutrophiles produits en réponse au pegfilgrastim possèdent des fonctions normales ou activées démontrées par les tests de chimiotactisme et de phagocytose. Comme pour d'autres facteurs de croissance hématopoïétiques, le G-CSF a montré in vitro des propriétés stimulantes des cellules endothéliales humaines. Le G-CSF peut promouvoir la croissance des cellules myéloïdes, dont celle des cellules malignes, in vitro et des effets similaires ont pu être observés sur certaines cellules non myéloïdes in vitro.
Dans deux études pivots randomisées, en double aveugle, chez des patientes atteintes d'un cancer du sein à haut risque de stade II-IV, traitées par une chimiothérapie myélosuppressive associant doxorubicine et docétaxel, l'administration de pegfilgrastim, à la posologie d'une injection unique une fois par cycle, a entraîné la réduction de la durée de la neutropénie et de l'incidence de la neutropénie fébrile de façon similaire à celle observée après administration quotidienne de filgrastim (avec une durée médiane d'administration de 11 jours). En l'absence de facteurs de croissance, une neutropénie de grade 4 d'une durée moyenne de 5 à 7 jours et une incidence de 30 % à 40 % de la neutropénie fébrile ont été décrites avec ce protocole. Dans une étude (n = 157), avec une dose unique de 6 mg de pegfilgrastim, la durée moyenne de neutropénie de grade 4 pour le groupe pegfilgrastim a été de 1,8 jour comparée à 1,6 jour pour le groupe filgrastim (différence de 0,23 jour ; IC à 95 % de - 0,15 à 0,63). Sur l'ensemble de l'étude, le taux de neutropénie fébrile a été de 13 % pour les patientes traitées par pegfilgrastim comparé à 20 % pour les patientes traitées par filgrastim (différence de 7 % ; IC à 95 % de - 19 % à 5 %). Dans une seconde étude (n = 310), avec une dose ajustée au poids du patient (100 µg/kg), la durée moyenne de la neutropénie de grade 4 pour le groupe pegfilgrastim a été de 1,7 jour comparée à 1,8 jour pour le groupe filgrastim (différence de 0,03 jour ; IC à 95 % de - 0,36 à 0,30). Le taux global de neutropénie fébrile a été de 9 % chez les patientes traitées par pegfilgrastim et de 18 % chez celles traitées par filgrastim (différence de 9 % ; IC à 95 % de - 16,8 % à - 1,1 %).
Dans une étude contrôlée contre placebo, en double aveugle, chez des patientes atteintes d'un cancer du sein, l'effet de pegfilgrastim sur l'incidence de la neutropénie fébrile a été évalué après administration d'un protocole de chimiothérapie associé à un taux de neutropénie fébrile de 10 à 20 % (docétaxel 100 mg/m2 toutes les 3 semaines pendant 4 cycles). 928 patientes ont été randomisées afin de recevoir, soit une dose unique de pegfilgrastim, soit le placebo, environ 24 heures (jour 2) après chaque cycle de chimiothérapie. L'incidence de la neutropénie fébrile a été plus faible chez les patientes du groupe pegfilgrastim comparés à celles du groupe placebo (1 % versus 17 %, p < 0,001). L'incidence des hospitalisations et de l'utilisation des anti-infectieux en IV, associées à un diagnostic clinique de neutropénie fébrile, a été plus faible pour le groupe pegfilgrastim comparé au groupe placebo (1 % versus 14 %, p < 0,001 ; et 2 % versus 10 %, p < 0,001).
Une petite étude (n = 83) randomisée de phase II, en double aveugle, menée chez des patients atteints de leucémie aiguë myéloïde de novo et traités par chimiothérapie, a comparé le pegfilgrastim (à la dose unique de 6 mg) au filgrastim, administrés lors de la chimiothérapie d'induction. Le temps médian de récupération d'une neutropénie sévère a été estimé à 22 jours dans les deux groupes traités. L'effet à long terme n'a pas été étudié (cf Mises en garde et Précautions d'emploi).
Dans une étude de phase II (n = 37) multicentrique, randomisée, en ouvert, chez des enfants présentant un sarcome et ayant reçu une dose de pegfilgrastim de 100 µg/kg après un premier cycle de chimiothérapie associant vincristine, doxorubicine et cyclophosphamide (VAdriaC/IE), la durée de la neutropénie sévère (neutrophiles < 0,5 x 109) était plus longue chez les jeunes enfants âgés de 0 à 5 ans (8,9 jours) comparé aux enfants âgés de 6 à 11 ans et de 12 à 21 ans (6 jours et 3,7 jours, respectivement), et aux adultes. De plus une incidence plus élevée de la neutropénie fébrile a été observée chez les jeunes enfants âgés de 0 à 5 ans (75 %), comparé aux enfants âgés de 6 à 11 ans et de 12 à 21 ans (70 % et 33 %, respectivement), et aux adultes (cf Effets indésirables, Pharmacocinétique).
PHARMACOCINÉTIQUE |
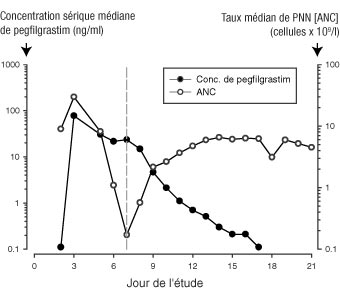
Après administration sous-cutanée unique de pegfilgrastim, le pic de concentration sérique apparaît entre 16 et 120 heures après l'injection et les concentrations sériques se maintiennent pendant la période de neutropénie qui suit la chimiothérapie myélosuppressive. L'élimination de pegfilgrastim n'est pas linéaire en fonction de la dose ; la clairance sérique de pegfilgrastim diminue lorsque les doses augmentent. Le pegfilgrastim semble s'éliminer principalement par la clairance neutrophile-dépendante qui est saturée à des doses plus élevées. La clairance étant autorégulée, la concentration sérique de pegfilgrastim diminue rapidement dès le début de la récupération en polynucléaires neutrophiles (PNN) : cf figure 1.
| Figure 1 : Concentration sérique médiane de pegfilgrastim et taux de polynucléaires neutrophiles (PNN) après une injection unique de 6 mg chez des patients traités par chimiothérapie |
 |
En raison du mécanisme de la clairance neutrophile-dépendante, la pharmacocinétique du pegfilgrastim ne devrait pas être modifiée par une insuffisance rénale ou hépatique. Dans une étude en ouvert après une injection unique (n = 31), l'insuffisance rénale à différents stades, y compris la maladie rénale à un stade avancé, n'a pas eu d'impact sur la pharmacocinétique du pegfilgrastim.
Des données limitées montrent que les paramètres pharmacocinétiques du pegfilgrastim ne sont pas modifiés chez les sujets âgés (> 65 ans).
- Enfant :
- La pharmacocinétique du pegfilgrastim a été étudiée chez 37 enfants atteints d'un sarcome et ayant reçu une dose de pegfilgrastim de 100 µg/kg après la fin d'une chimiothérapie (VAdriaC/IE). Les plus jeunes enfants (0 à 5 ans) ont présenté une exposition moyenne au pegfilgrastim (AUC) (± écart-type) (47,9 µg x h/ml ± 22,5) plus élevée que les enfants âgés de 6 à 11 ans et de 12 à 21 ans (22,0 µg x h/ml ± 13,1 et 29,3 µg x h/ml ± 23,2, respectivement) : cf Pharmacodynamie. A l'exception du groupe d'enfants les plus jeunes (0-5 ans), l'AUC moyenne chez les enfants semble similaire à celle des adultes présentant un cancer du sein à haut risque de stade II-IV et ayant reçu 100 µg/kg de pegfilgrastim après la fin d'une chimiothérapie par doxorubicine/docétaxel (cf Effets indésirables, Pharmacodynamie).
SÉCURITE PRÉCLINIQUE |
Les données d'études précliniques conventionnelles portant sur la toxicité à doses répétées ont mis en évidence les effets pharmacologiques attendus, tels qu'une augmentation du nombre de leucocytes, une hyperplasie myéloïde de la moelle osseuse, une hématopoïèse extramédullaire et une splénomégalie.
Aucun effet indésirable n'a été observé dans la progéniture de la rate ayant reçu du pegfilgrastim par voie sous-cutanée pendant la gestation ; par ailleurs, le pegfilgrastim administré à faibles doses par voie sous-cutanée a entraîné chez la lapine une toxicité embryofoetale (perte embryonnaire). Dans les études effectuées chez le rat, le passage transplacentaire du pegfilgrastim a été mis en évidence. Les conséquences de ces observations ne sont pas connues chez l'homme.
INCOMPATIBILITÉS |
Ce médicament ne doit pas être mélangé avec d'autres médicaments, particulièrement avec les solutions de chlorure de sodium.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 30 mois.
A conserver au réfrigérateur (entre + 2 °C et + 8 °C).
Neulasta peut supporter d'être exposé à température ambiante (sans dépasser + 30 °C) pendant une période unique maximale de 72 heures. Si Neulasta est laissé à température ambiante pendant plus de 72 heures, il doit être éliminé.
Ne pas congeler. Une congélation accidentelle pendant une période unique de moins de 24 heures n'affecte pas la stabilité de Neulasta.
Conserver le conditionnement primaire dans l'emballage extérieur à l'abri de la lumière.
MODALITÉS MANIPULATION/ÉLIMINATION |
Avant administration, la solution Neulasta doit être inspectée visuellement pour mettre en évidence l'absence de particules. Seule une solution limpide et incolore peut être injectée.
Une agitation excessive peut provoquer la formation d'agrégats de pegfilgrastim rendant la solution biologiquement inactive.
Laisser la seringue préremplie atteindre la température ambiante avant l'injection.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| Médicament soumis à prescription initiale hospitalière trimestrielle. | |
| AMM | EU/1/02/227/004 ; CIP 3400939535166 (RCP rév 08.04.2011). |
| Prix : | 1132.84 euros (1 seringue sécurisée). |
| Remb Séc soc à 100 %. Collect. | |
Titulaire de l'AMM : Amgen Europe B.V., Minervum 7061, 4817 ZK Breda, Pays-Bas.
AMGEN
62, bd Victor-Hugo. 92200 Neuilly-sur-Seine
Tél : 01 40 88 27 00. Fax : 01 40 88 27 90
Info médic et pharmacovigilance :
Tél : 09 69 36 33 63
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale
