ZOMETA®
acide zolédronique
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p flacon | |
| Acide zolédronique (DCI) monohydraté exprimé en acide zolédronique anhydre | 4 mg |
INDICATIONS |
- Prévention des complications osseuses (fractures pathologiques, compression médullaire, irradiation ou chirurgie osseuse, hypercalcémie induite par des tumeurs) chez des patients atteints de pathologie maligne à un stade avancé avec atteinte osseuse.
- Traitement de l'hypercalcémie induite par des tumeurs (TIH).
POSOLOGIE ET MODE D'ADMINISTRATION |
La solution à diluer de Zometa ne doit pas être mélangée avec des solutions contenant du calcium ou avec d'autres solutions pour perfusion contenant des cations divalents telle que la solution de Ringer lactate, et doit être administrée par voie de perfusion séparée en solution intraveineuse unique.
- Prévention des complications osseuses chez des patients atteints de pathologie maligne à un stade avancé avec atteinte osseuse :
-
- Adulte et sujet âgé :
- La dose recommandée dans la prévention des complications osseuses chez des patients atteints de pathologie maligne à un stade avancé avec atteinte osseuse est de 4 mg d'acide zolédronique. La solution doit être diluée dans 100 ml de solution stérile de chlorure de sodium à 0,9 % m/v ou de solution de glucose à 5 % m/v, et administrée par perfusion intraveineuse d'une durée d'au moins 15 minutes toutes les 3 à 4 semaines.
- Les patients devront aussi recevoir, par voie orale, un apport de 500 mg de calcium et de 400 UI de vitamine D par jour.
- Coût du traitement journalier : 11,24 à 14,99 euro(s).
- Traitement de l'hypercalcémie induite par des tumeurs :
-
- Adulte et sujet âgé :
- La dose recommandée dans l'hypercalcémie (calcémie corrigée en fonction de l'albumine >= 12,0 mg/dl ou 3,0 mmol/l) est de 4 mg d'acide zolédronique. La solution doit être diluée dans 100 ml de solution stérile de chlorure de sodium à 0,9 % m/v ou de glucose à 5 % m/v, et administrée par perfusion intraveineuse unique d'au moins 15 minutes. Les patients doivent être correctement hydratés avant et après l'administration de Zometa.
- Coût d'un flacon : 314,76 euro(s).
- Insuffisance rénale :
-
- Hypercalcémie induite par des tumeurs (TIH) :
- Le traitement par Zometa des patients ayant une hypercalcémie induite par des tumeurs et présentant également une atteinte rénale sévère devra être envisagé uniquement après l'évaluation des risques et des bénéfices de ce traitement. Dans les études cliniques, les patients ayant une créatininémie > 400 µmol/l ou > 4,5 mg/dl ont été exclus. Aucune adaptation de la dose n'est nécessaire chez les patients présentant une hypercalcémie induite par des tumeurs avec une créatininémie < 400 µmol/l ou < 4,5 mg/dl (cf Mises en garde et Précautions d'emploi).
-
- Prévention des complications osseuses chez des patients atteints de pathologie maligne à un stade avancé avec atteinte osseuse :
- A l'initiation du traitement par Zometa des patients avec un myélome multiple ou avec atteintes osseuses métastatiques secondaires à des tumeurs solides, la créatininémie et la clairance à la créatinine (Clcr) devront être évaluées. La Clcr est calculée selon la formule de Cockcroft-Gault à partir de la créatininémie. Zometa n'est pas recommandé chez des patients présentant une atteinte rénale sévère avant l'initiation du traitement, atteinte rénale qui est définie par une Clcr < 30 ml/min pour cette population. Dans les études cliniques menées avec Zometa, les patients ayant une créatininémie > 265 µmol/l ou 3,0 mg/dl étaient exclus.
- Chez les patients avec des métastases osseuses présentant une atteinte rénale légère à modérée avant l'initiation du traitement, atteinte rénale qui est définie par une Clcr de 30 à 60 ml/min, la dose recommandée de Zometa est la suivante (cf Mises en garde et Précautions d'emploi) :
-
Clairance à la créatinine initiale (ml/min) Dose recommandée de Zometa* > 60 4,0 mg 50-60 3,5 mg* 40-49 3,3 mg* 30-39 3,0 mg* -
*
Les doses ont été calculées en vue d'atteindre une valeur de l'ASC de 0,66 mg x h/l (pour une Clcr = 75 ml/min), l'objectif étant que, chez les patients avec atteinte rénale, les doses réduites de Zometa permettent d'obtenir la même ASC que celle observée chez des patients avec une clairance à la créatinine de 75 ml/min.
- Après l'initiation du traitement, la créatininémie devra être mesurée avant chaque administration de Zometa et le traitement devra être suspendu si la fonction rénale s'est détériorée.
- Dans les études cliniques, l'altération de la fonction rénale était définie comme suit :
- une augmentation de 0,5 mg/dl ou 44 µmol/l chez les patients qui avaient une valeur de la créatinine de base normale (< 1,4 mg/dl ou < 124 µmol/l) ;
- une augmentation de 1,0 mg/dl ou 88 µmol/l chez les patients qui avaient une valeur de la créatinine de base anormale (> 1,4 mg/dl ou > 124 µmol/l).
- une augmentation de 0,5 mg/dl ou 44 µmol/l chez les patients qui avaient une valeur de la créatinine de base normale (< 1,4 mg/dl ou < 124 µmol/l) ;
- Dans les études cliniques, le traitement par Zometa était repris uniquement lorsque la valeur de la créatininémie était revenue à la valeur de base ± 10 % (cf Mises en garde et Précautions d'emploi). Le traitement par Zometa devra être repris à la même dose que celle administrée avant l'interruption du traitement.
-
- Instructions pour préparer les doses réduites de Zometa :
- Prélever un volume approprié de la solution concentrée comme suit :
- 4,4 ml pour une dose de 3,5 mg ;
- 4,1 ml pour une dose de 3,3 mg ;
- 3,8 ml pour une dose de 3,0 mg ;
- 4,4 ml pour une dose de 3,5 mg ;
- Pour toute information concernant la dilution de Zometa, se reporter à la rubrique Modalités Manipulation/Élimination.
- La quantité prélevée de la solution concentrée doit ensuite être diluée dans 100 ml de solution stérile de chlorure de sodium à 0,9 % m/v ou de solution de glucose à 5 % m/v. La dose doit être administrée en perfusion intraveineuse d'une durée d'au moins 15 minutes.
- Enfant :
- L'utilisation de Zometa chez l'enfant a été étudiée dans 2 études cliniques dans le traitement de l'ostéogenèse imparfaite sévère (cf Pharmacodynamie). Zometa ne doit pas être utilisé chez l'enfant étant donné que la sécurité d'emploi et l'efficacité n'ont pas été établies chez l'enfant (cf Mises en garde et Précautions d'emploi, Pharmacodynamie).
CONTRE-INDICATIONS |
- Hypersensibilité à la substance active, à d'autres bisphosphonates ou à l'un des excipients contenus dans Zometa.
- Allaitement : cf Grossesse et Allaitement.
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
- Générales :
- Les patients devront être évalués avant l'administration de Zometa pour s'assurer qu'ils sont correctement hydratés.
- L'hyperhydratation doit être évitée chez les patients présentant un risque d'insuffisance cardiaque.
- Les paramètres métaboliques standards associés à l'hypercalcémie, tels que la calcémie, la phosphatémie et la magnésémie, doivent être surveillés avec attention après initiation du traitement par Zometa.
- En cas d'hypocalcémie, d'hypophosphatémie ou d'hypomagnésémie, un traitement de supplémentation de courte durée peut être nécessaire.
- Les patients ayant une hypercalcémie non traitée présentent généralement une atteinte de la fonction rénale ; il est donc recommandé de surveiller avec précaution la fonction rénale.
- La substance active contenue dans Zometa est identique à celle d'Aclasta (acide zolédronique). Les patients traités par Zometa ne doivent pas être traités par Aclasta de manière concomitante.
- La sécurité d'emploi et l'efficacité de Zometa n'ont pas été établies chez l'enfant (cf Pharmacodynamie).
- Insuffisance rénale :
- Les patients ayant une TIH et présentant une altération de la fonction rénale devront être évalués de façon appropriée pour apprécier le rapport bénéfice/risque du traitement avec Zometa.
- La décision de traiter les patients ayant des métastases osseuses afin de prévenir les complications osseuses devra être prise en tenant compte du fait que l'effet du traitement commence à s'observer au bout de 2 à 3 mois.
- Comme avec les autres bisphosphonates, Zometa a été associé à des cas de dysfonctionnements rénaux. Les facteurs qui peuvent augmenter le risque d'une altération de la fonction rénale comprennent la déshydratation, l'insuffisance rénale préexistante, les cycles multiples de Zometa et d'autres bisphosphonates, aussi bien que d'autres médicaments néphrotoxiques. Bien que le risque soit réduit en administrant sur 15 minutes la dose de 4 mg de Zometa, une altération de la fonction rénale peut cependant se produire. Une altération rénale, une progression de l'insuffisance rénale et un cas de dialyse ont été rapportés chez des patients après une dose initiale ou une seule dose de Zometa. Des augmentations de la créatininémie peuvent aussi s'observer, quoique moins fréquemment, chez quelques patients qui reçoivent Zometa en administration chronique aux doses recommandées pour la prévention des complications osseuses.
- Les patients devront avoir un dosage de leur créatininémie avant chaque administration de Zometa. En cas d'initiation de traitement par Zometa chez des patients ayant des métastases osseuses et une atteinte rénale légère à modérée, des doses plus faibles de Zometa sont recommandées. En cas d'altération de la fonction rénale au cours du traitement, Zometa devra être interrompu. Zometa devra être repris uniquement lorsque la créatininémie est revenue à la valeur de base ± 10 % (cf Posologie et Mode d'administration).
- En raison de l'effet potentiel des bisphosphonates (y compris Zometa) sur la fonction rénale, du manque de données sur la tolérance clinique chez des patients ayant avant traitement une atteinte rénale sévère (définie dans les études cliniques par une créatininémie >= 400 µmol/l ou >= 4,5 mg/dl chez des patients ayant une TIH et par une créatininémie >= 265 µmol/l ou >= 3,0 mg/dl chez des patients atteints de pathologie maligne avec atteinte osseuse) et compte tenu des données pharmacocinétiques encore limitées chez les patients ayant au départ une atteinte rénale sévère (clairance de la créatinine < 30 ml/min), l'utilisation de Zometa n'est pas recommandée chez des patients ayant une atteinte rénale sévère.
- Insuffisance hépatique :
- Les données cliniques disponibles sont limitées chez les patients ayant une insuffisance hépatique sévère, aussi aucune recommandation spécifique ne peut être donnée chez cette population de patients.
- Ostéonécrose de la mâchoire :
- Une ostéonécrose de la mâchoire a été rapportée chez des patients, principalement chez ceux atteints d'un cancer et traités par des bisphosphonates, y compris Zometa. La plupart de ces patients recevaient aussi une chimiothérapie et des corticoïdes. La majorité des cas rapportés ont été associés à des interventions dentaires telles qu'une extraction dentaire. Plusieurs présentaient des signes d'infection localisée, y compris une ostéomyélite.
- Un examen dentaire avec des soins dentaires préventifs appropriés devra être pris en considération avant l'instauration d'un traitement par bisphosphonates chez des patients présentant des facteurs de risque associés (par exemple : cancer, chimiothérapie, corticoïdes ou mauvaise hygiène buccale).
- Au cours du traitement, ces patients devront éviter dans la mesure du possible toutes interventions dentaires invasives. Pour les patients qui développent une ostéonécrose de la mâchoire au cours d'un traitement par bisphosphonates, une chirurgie dentaire peut aggraver cette atteinte. Pour les patients nécessitant une intervention dentaire, il n'y a pas de donnée disponible suggérant que l'arrêt du traitement par bisphosphonates diminuerait le risque d'ostéonécrose de la mâchoire. L'appréciation clinique du médecin traitant devrait orienter la prise en charge de chaque patient en se basant sur l'évaluation individuelle du rapport bénéfice/risque.
- Douleurs musculosquelettiques :
- Après mise sur le marché, des douleurs osseuses, articulaires et/ou musculaires sévères ou occasionnellement invalidantes ont été rapportées chez des patients traités par bisphosphonates. Toutefois, de tels cas n'ont été rapportés que peu fréquemment. Zometa (l'acide zolédronique) appartient à cette classe de médicaments. Le délai d'apparition des symptômes varie d'un jour à plusieurs mois après le début du traitement. Chez la majorité des patients, ces symptômes ont régressé après l'arrêt du traitement. Une réapparition des symptômes a été observée chez certains patients après la reprise du traitement avec le même médicament ou avec un autre bisphosphonate.
INTERACTIONS |
In vitro, l'acide zolédronique ne présente pas de liaison notable aux protéines plasmatiques et n'inhibe pas les enzymes du cytochrome P450 humaines (cf Pharmacocinétique), mais aucune étude clinique d'interaction proprement dite n'a été menée. La prudence est conseillée lorsque les bisphosphonates sont administrés avec des aminosides, puisque les deux substances peuvent avoir un effet additif, entraînant un taux plus faible de la calcémie sur des périodes plus longues que celles requises. La prudence est requise lorsque Zometa est administré avec d'autres substances potentiellement néphrotoxiques. Il faut aussi prêter attention à la survenue possible d'une hypomagnésémie pendant le traitement.
Chez les patients atteints de myélome multiple, le risque d'altération de la fonction rénale peut être augmenté lorsque les bisphosphonates sont utilisés par voie intraveineuse en association avec la thalidomide.
GROSSESSE et ALLAITEMENT |
Il n'existe pas de données suffisantes concernant l'utilisation de l'acide zolédronique chez la femme enceinte. Des études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction avec l'acide zolédronique (cf Sécurité préclinique). Le risque potentiel en clinique n'est pas connu. Zometa ne doit pas être utilisé pendant la grossesse.
Allaitement :
Il n'est pas établi si l'acide zolédronique est excrété dans le lait maternel. Zometa est contre-indiqué chez la femme qui allaite (cf Contre-indications).
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
Fréquemment, la réduction de l'excrétion rénale du calcium est accompagnée d'une baisse asymptomatique de la phosphatémie (chez environ 20 % des patients), ne nécessitant pas de traitement. La calcémie peut être abaissée à des valeurs d'hypocalcémie asymptomatique chez environ 3 % des patients.
Des effets indésirables gastro-intestinaux, tels que des nausées (5,8 %) et des vomissements (2,6 %) ont été rapportés après une perfusion intraveineuse de Zometa. Occasionnellement, des réactions locales au point d'injection telles que rougeur ou oedème et/ou douleurs ont aussi été observées chez moins de 1 % des patients.
Une anorexie a été rapportée chez 1,5 % des patients traités par Zometa 4 mg.
Peu de cas d'éruption cutanée ou de prurit ont été observés (moins de 1 %).
Comme avec d'autres bisphosphonates, des cas de conjonctivite, chez approximativement 1 % des patients, ont été rapportés.
Il a été rapporté des cas d'altération de la fonction rénale (2,3 %), bien que l'étiologie semble être multifactorielle dans de nombreux cas.
Sur la base d'une analyse groupée des études contrôlées versus placebo, une anémie sévère (Hb < 8,0 g/dl) a été rapportée chez 5,2 % des patients ayant reçu Zometa versus 4,2 % des patients ayant reçu le placebo.
Les réactions indésirables suivantes, énumérées dans le tableau 1, ont été collectées dans des études cliniques et principalement après l'administration chronique du traitement par l'acide zolédronique.
Tableau 1 : Les réactions indésirables sont classées par ordre de fréquence décroissante en utilisant la convention suivante : très fréquent (>= 1/10) ; fréquent (>= 1/100, < 1/10) ; peu fréquent (>= 1/1000, < 1/100) ; rare (>= 1/10 000, < 1/1000) ; très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
| Classe-organe | Effets indésirables |
| Affections hématologiques et du système lymphatique | Fréquent : anémie |
| Peu fréquent : thrombopénie, leucopénie | |
| Rare : pancytopénie | |
| Affections du système nerveux | Fréquent : céphalée |
| Peu fréquent : étourdissements, paresthésie, trouble du goût, hypoesthésie, hyperesthésie, tremblements | |
| Affections psychiatriques | Peu fréquent : anxiété, troubles du sommeil |
| Rare : confusion | |
| Affections oculaires | Fréquent : conjonctivite |
| Peu fréquent : vision trouble | |
| Très rare : uvéite, épisclérite | |
| Affections gastro-intestinales | Fréquent : nausées, vomissements, anorexie |
| Peu fréquent : diarrhée, constipation, douleurs abdominales, dyspepsie, stomatite, bouche sèche | |
| Affections respiratoires, thoraciques et médiastinales | Peu fréquent : dyspnée, toux |
| Affections de la peau et du tissu sous-cutané | Peu fréquent : prurit, éruptions cutanées (y compris éruptions érythémateuses et maculaires), transpiration accrue |
| Affections musculosquelettiques et systémiques | Fréquent : douleurs osseuses, myalgie, arthralgie, douleur généralisée |
| Peu fréquent : crampes musculaires | |
| Affections cardiaques | Peu fréquent : hypertension, hypotension |
| Rare : bradycardie | |
| Affections du rein et des voies urinaires | Fréquent : atteintes rénales |
| Peu fréquent : insuffisance rénale aiguë, hématurie, protéinurie | |
| Affections du système immunitaire | Peu fréquent : réaction d'hypersensibilité |
| Rare : oedème de Quincke (angioneurotique) | |
| Troubles généraux et anomalies au site d'administration | Fréquent : fièvre, syndrome pseudogrippal (y compris fatigue, frissons, malaise et bouffée vasomotrice) |
| Peu fréquent : asthénie, oedème périphérique, réactions au site d'injection (y compris douleurs, irritation, tuméfaction, induration), douleur thoracique, prise de poids | |
| Investigations | Très fréquent : hypophosphatémie |
| Fréquent : augmentation de la créatininémie et de l'uricémie, hypocalcémie | |
| Peu fréquent : hypomagnésémie, hypokaliémie | |
| Rare : hyperkaliémie, hypernatrémie |
- Expérience depuis la commercialisation :
- Les effets indésirables suivants ont été rapportés depuis la commercialisation de Zometa.
- Des cas d'ostéonécroses (principalement de la mâchoire) ont été rapportés, principalement chez des patients atteints d'un cancer et traités par des bisphosphonates, y compris Zometa. Beaucoup de ces patients présentaient des signes d'infection locale, y compris une ostéomyélite et la majorité des cas concernait des patients atteints d'un cancer et ayant subi une extraction dentaire ou d'autres chirurgies dentaires. L'ostéonécrose de la mâchoire présente de multiples facteurs de risque bien documentés incluant le diagnostic d'un cancer, les traitements associés (par exemple : chimiothérapie, radiothérapie, corticothérapie) et des affections associées (par exemple : anémie, troubles de la coagulation, infection, maladie buccale préexistante). Bien que la causalité n'ait pas été établie, il est prudent d'éviter une chirurgie dentaire dont la guérison pourrait être retardée (cf Mises en garde et Précautions d'emploi).
- Dans de très rares cas, les effets suivants ont été rapportés : hypotension entraînant une syncope ou un collapsus cardio-vasculaire principalement chez des patients avec des facteurs de risques sous-jacents, fibrillation auriculaire, somnolence, bronchoconstriction, réaction ou choc anaphylactique, urticaire, sclérite et inflammation oculaire. Ces cas rapportés étant issus d'une population de taille incertaine et étant sujets à de multiples facteurs, il est difficile d'évaluer leur causalité et d'estimer le taux d'incidence de ces événements.
- Enfants :
- Les données de sécurité d'emploi chez l'enfant sont résumées en rubrique Pharmacodynamie.
SURDOSAGE |
PHARMACODYNAMIE |
Classe pharmacothérapeutique : bisphosphonates (code ATC : M05BA08).
L'acide zolédronique appartient à la classe des bisphosphonates et agit principalement sur l'os. Il inhibe la résorption ostéoclastique osseuse.
L'action sélective des bisphosphonates sur l'os découle de leur forte affinité pour l'os minéralisé, mais le mécanisme moléculaire précis, menant à l'inhibition de l'activité ostéoclastique, n'est pas encore élucidé. Dans les études à long terme menées chez l'animal, l'acide zolédronique inhibe la résorption osseuse sans effet défavorable sur la formation, la minéralisation ou les propriétés mécaniques de l'os.
En plus d'être un puissant inhibiteur de la résorption osseuse, l'acide zolédronique possède également plusieurs propriétés antitumorales qui pourraient contribuer à son efficacité globale dans le traitement de la maladie métastatique osseuse. Les propriétés suivantes ont été démontrées dans des études précliniques :- In vivo : inhibition de la résorption ostéoclastique osseuse qui altère le microenvironnement médullaire, le rendant moins favorable à la croissance des cellules tumorales, activité antiangiogénique et activité antalgique.
- In vitro : inhibition de la prolifération ostéoblastique, activité cytostatique directe et pro-apoptotique sur les cellules tumorales, effet cytostatique synergique en association à d'autres médicaments anticancéreux, activité antiadhésion/invasion cellulaire.
- Résultats des études cliniques dans la prévention des complications osseuses chez des patients atteints de pathologie maligne à un stade avancé avec atteinte osseuse :
- La première étude randomisée, en double aveugle, contrôlée versus placebo comparait Zometa au placebo dans la prévention des complications osseuses (Skeletal Related Events : SREs) chez des patients présentant un cancer de la prostate. Zometa 4 mg a réduit significativement la proportion de patients présentant au moins une complication osseuse (SRE), a retardé le délai médian de survenue de la première complication osseuse de plus de 5 mois et a réduit l'incidence annuelle de complications osseuses par patient (taux de morbidité osseuse). L'analyse des « événements multiples » a montré une réduction de 36 % du risque de développer des SREs dans le groupe Zometa en comparaison avec le groupe placebo. Les patients ayant reçu Zometa ont rapporté moins d'augmentation de la douleur que ceux ayant reçu le placebo, avec des différences significatives à 3, 9, 21 et 24 mois. Il y a eu moins de patients traités par Zometa qui ont souffert de fractures pathologiques. Les effets du traitement étaient moins prononcés chez les patients présentant des lésions blastiques. Les résultats d'efficacité sont rapportés dans le tableau 2.
- Dans une seconde étude comprenant des tumeurs solides autres que le cancer du sein ou le cancer de la prostate, Zometa 4 mg a réduit significativement la proportion de patients avec au moins une SRE, a retardé le délai médian de survenue de la première complication osseuse de plus de 2 mois et a réduit le taux de morbidité osseuse. L'analyse des « événements multiples » a montré une réduction de 30,7 % du risque de développer des SREs dans le groupe Zometa en comparaison avec le placebo. Les résultats d'efficacité sont rapportés dans le tableau 3.
-
Tableau 2 : Résultats d'efficacité (patients présentant un cancer de la prostate et recevant une hormonothérapie) : Toute complication osseuse (TIH incluse) Fractures* Radiothérapie osseuse Zometa 4 mg Placebo Zometa 4 mg Placebo Zometa 4 mg Placebo N 214 208 214 208 214 208 Proportion de patients avec complications osseuses (%) 38 49 17 25 26 33 Valeur p 0,028 0,052 0,119 Médiane de survenue de la 1re complication osseuse (jours) 488 321 NA(1) NA(1) NA(1) 640 Valeur p 0,009 0,020 0,055 Taux de morbidité osseuse 0,77 1,47 0,20 0,45 0,42 0,89 Valeur p 0,005 0,023 0,060 Réduction du risque de développer des complications osseuses (analyse des « événements multiples »)** (%) 36 NApp(2) NApp(2) NApp(2) NApp(2) Valeur p 0,002 NApp(2) NApp(2) -
*
incluant les fractures vertébrales et non vertébrales
-
**
prend en compte toutes les complications osseuses, aussi bien le nombre total que la durée entre chaque complication au cours de l'étude
-
(1)
NA = non atteint
-
(2)
NApp = non applicable
-
Tableau 3 : Résultats d'efficacité (patients présentant des tumeurs solides autres que cancer du sein ou cancer de la prostate) : Toute complication osseuse (TIH incluse) Fractures* Radiothérapie osseuse Zometa 4 mg Placebo Zometa 4 mg Placebo Zometa 4 mg Placebo N 257 250 257 250 257 250 Proportion de patients avec complications osseuses (%) 39 48 16 22 29 34 Valeur p 0,039 0,064 0,173 Médiane de survenue de la 1re complication osseuse (jours) 236 155 NA(1) NA(1) 424 307 Valeur p 0,009 0,020 0,079 Taux de morbidité osseuse 1,74 2,71 0,39 0,63 1,24 1,89 Valeur p 0,012 0,066 0,099 Réduction du risque de développer des complications osseuses (analyse des « événements multiples »)** (%) 30,7 NApp(2) NApp(2) NApp(2) NApp(2) Valeur p 0,003 NApp(2) NApp(2) -
*
incluant les fractures vertébrales et non vertébrales
-
**
prend en compte toutes les complications osseuses, aussi bien le nombre total que la durée entre chaque complication au cours de l'étude
-
(1)
NA = non atteint
-
(2)
NApp = non applicable
- Dans une troisième étude de phase III, randomisée, en double aveugle, Zometa 4 mg a été comparé à 90 mg de pamidronate administrés toutes les 3 à 4 semaines chez des patients ayant un myélome multiple ou un cancer du sein avec au moins une complication osseuse. Les résultats ont démontré que Zometa 4 mg avait une efficacité comparable à 90 mg de pamidronate dans la prévention des complications osseuses. L'analyse des « événements multiples » a montré une réduction significative de 16 % du risque de développer des complications osseuses chez les patients traités par Zometa 4 mg en comparaison avec ceux traités par le pamidronate. Les résultats d'efficacité sont rapportés dans le tableau 4.
-
Tableau 4 : Résultats d'efficacité (patients présentant un cancer du sein ou un myélome multiple) : Toute complication osseuse (TIH incluse) Fractures* Radiothérapie osseuse Zometa 4 mg Pam 90 mg Zometa 4 mg Pam 90 mg Zometa 4 mg Pam 90 mg N 561 555 561 555 561 555 Proportion de patients avec complications osseuses (%) 48 52 37 39 19 24 Valeur p 0,198 0,653 0,037 Médiane de survenue de la 1re complication osseuse (jours) 376 356 NA(1) 714 NA(1) NA(1) Valeur p 0,151 0,672 0,026 Taux de morbidité osseuse 1,04 1,39 0,53 0,60 0,47 0,71 Valeur p 0,084 0,614 0,015 Réduction du risque de développer des complications osseuses (analyse des « événements multiples »)** (%) 16 NApp(2) NApp(2) NApp(2) NApp(2) Valeur p 0,030 NApp(2) NApp(2) -
*
incluant les fractures vertébrales et non vertébrales
-
**
prend en compte toutes les complications osseuses, aussi bien le nombre total que la durée entre chaque complication au cours de l'étude
-
(1)
NA = non atteint
-
(2)
NApp = non applicable
- Zometa a aussi été étudié dans une étude randomisée, en double-aveugle, contrôlée versus placebo, chez 228 patients avec métastases osseuses documentées provenant d'un cancer du sein, pour évaluer l'effet de Zometa sur l'incidence des complications osseuses (Skeletal Related Events : SREs), déterminée comme le rapport du nombre total de SREs (excluant l'hypercalcémie et ajusté en fonction des fractures antérieures) sur le temps d'exposition au risque. Les patients recevaient 4 mg de Zometa ou du placebo toutes les 4 semaines pendant une année. La répartition des patients entre le groupe traité par Zometa et le groupe placebo était homogène.
- L'incidence des SREs (complication/année-patient) était de 0,628 pour Zometa et 1,096 pour le placebo. La proportion de patients avec au moins une SRE (excluant l'hypercalcémie) était de 29,8 % dans le groupe traité par Zometa versus 49,6 % pour le groupe placebo (p = 0,003). Le temps médian d'apparition de la première SRE n'était pas encore atteint dans le groupe Zometa à la fin de l'étude et était significativement prolongé par comparaison au placebo (p = 0,007). Dans une analyse d'événements multiples, Zometa a réduit le risque de SREs de 41 % par comparaison au placebo (risque relatif = 0,59 ; p = 0,019).
- Dans le groupe Zometa, une amélioration statistiquement significative du score de douleur (utilisation de la « Brief Pain Inventory » [BPI]) a été observée à 4 semaines et à tous les moments ultérieurs de l'étude, en comparaison au placebo (figure 1). Le score de douleur avec Zometa était constamment inférieur à celui observé à l'état initial et il était accompagné d'une tendance à la réduction du score d'analgésie.
-
Figure 1 : Variations moyennes du score de douleur BPI par rapport à l'état initial. Pour la comparaison des traitements (Zometa versus placebo), les différences statistiquement significatives sont annotées d'un astérisque (* p < 0,05). 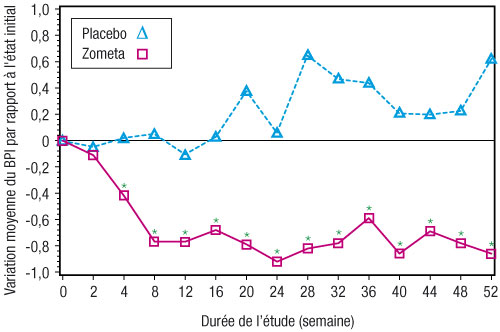
- Résultats des études cliniques dans le traitement des hypercalcémies induites par des tumeurs (TIH) :
- Les études cliniques dans l'hypercalcémie induite par des tumeurs (TIH) ont démontré que l'effet de l'acide zolédronique est caractérisé par une baisse de la calcémie et de l'excrétion urinaire de calcium. Dans les études phase I de recherche de doses, chez des patients présentant une hypercalcémie légère à modérée secondaire à des tumeurs (TIH), les doses efficaces testées ont été approximativement de 1,2 à 2,5 mg.
- Pour évaluer les effets de Zometa par rapport à 90 mg de pamidronate, les résultats de deux études pivots multicentriques chez des patients ayant une TIH ont été combinés dans une analyse préalablement programmée. Il a été observé une normalisation de la calcémie corrigée plus rapide au 4e jour avec 8 mg de Zometa et au 7e jour avec 4 mg et 8 mg de Zometa. Les taux de réponse suivants ont été observés :
-
Tableau 5 : Pourcentage de patients ayant une réponse complète par jour dans les études TIH combinées : 4e jour 7e jour 10e jour Zometa 4 mg (n = 86) 45,3 %
(p = 0,104)82,6 %
(p = 0,005)*88,4 %
(p = 0,002)*Zometa 8 mg (n = 90) 55,6 %
(p = 0,021)*83,3 %
(p = 0,010)*86,7 %
(p = 0,015)*Pamidronate 90 mg (n = 99) 33,3 % 63,6 % 69,7 % -
*
valeurs p comparées au pamidronate
- Le délai médian de normalisation de la calcémie a été de 4 jours. Le délai médian avant la rechute (réaugmentation de la calcémie corrigée en fonction de l'albumine >= 2,9 mmol/l) a été de 30 à 40 jours pour les patients traités par Zometa versus 17 jours pour ceux traités par 90 mg de pamidronate (p = 0,001 pour 4 mg et p = 0,007 pour 8 mg). Il n'y a pas de différence statistiquement significative entre les deux doses de Zometa.
- Dans les études cliniques, 69 patients qui ont rechuté ou qui étaient réfractaires au traitement initial (Zometa 4 mg, 8 mg ou pamidronate 90 mg) ont été traités une seconde fois avec Zometa 8 mg. Le taux de réponse chez ces patients était environ de 52 %. Puisque ces patients ont été traités de nouveau uniquement par Zometa 8 mg, il n'y a pas de données disponibles qui permettent de comparer avec la dose de 4 mg.
- Dans les études cliniques réalisées chez des patients avec hypercalcémie induite par des tumeurs, le profil global de tolérance dans les trois groupes de traitement (acide zolédronique 4 mg et 8 mg et pamidronate 90 mg) était similaire en nature et en sévérité.
- Enfants :
-
- Résultats des études cliniques dans le traitement de l'ostéogenèse imparfaite sévère chez les enfants âgés de 1 à 17 ans :
- Les effets de l'acide zolédronique administré par voie intraveineuse chez l'enfant (âgé de 1 à 17 ans) atteint d'ostéogenèse imparfaite sévère (types I, III, et IV) ont été comparés avec le pamidronate administré par voie intraveineuse dans une étude internationale, multicentrique, randomisée et en ouvert avec respectivement 74 et 76 patients dans chaque groupe de traitement. La durée du traitement était de 12 mois précédée d'une période de sélection de 4 à 9 semaines pendant laquelle une supplémentation en vitamine D et en calcium a été administrée pendant au moins 2 semaines. Dans le programme clinique, les patients âgés de 1 à 3 ans recevaient 0,025 mg/kg d'acide zolédronique (jusqu'à une dose unique maximale de 0,35 mg) tous les 3 mois et les patients âgés de 3 à 17 ans recevaient 0,05 mg/kg d'acide zolédronique (jusqu'à une dose unique maximale de 0,83 mg) tous les 3 mois. Une étude d'extension a été menée afin d'examiner la tolérance générale et rénale à long terme de l'acide zolédronique une ou deux fois par an sur une période d'extension de 12 mois de traitement chez les enfants ayant reçu un an de traitement soit par l'acide zolédronique soit par le pamidronate dans l'étude principale.
- L'objectif principal de l'étude était le pourcentage de changement de la densité minérale osseuse (DMO) au col fémoral après 12 mois de traitement. Les effets estimés des traitements sur la DMO étaient similaires mais le design de l'essai n'était pas suffisamment robuste pour établir la non infériorité d'efficacité de Zometa. En particulier, il n'était pas clairement démontré l'efficacité sur des fractures ou sur la douleur. Des fractures des os longs des extrémités inférieurs ont été rapportés chez approximativement 24 % (fémur) et 14 % (tibia) des patients traités par l'acide zolédronique contre 12 % et 5 % des patients traités par le pamidronate atteints d'ostéogenèse imparfaite sévère, sans tenir compte du type de maladie et de la causalité mais l'incidence moyenne des fractures était comparable chez les patients traités par l'acide zolédronique et chez ceux traités par le pamidronate : 43 % (32/74) contre 41 % (31/76). L'interprétation du risque de fracture est compromis par le fait que les fractures sont des événements indésirables fréquents chez les patients atteints d'ostéogenèse imparfaite sévère, du fait de la maladie.
- Le type d'événements indésirables observés dans cette population était généralement similaire avec ceux précédemment observés chez les adultes ayant un cancer des os avancé (cf Effets indésirables). Les réactions indésirables sont classées par ordre de fréquence décroissante en utilisant la convention suivante : très fréquent (>= 1/10), fréquent (>= 1/100, < 1/10), peu fréquent (>= 1/1000, < 1/100), rare (>= 1/10 000, < 1/1000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
-
Tableau 6 : Réactions indésirables observées chez l'enfant atteint d'ostéogenèse imparfaite sévère. Affections du système nerveux : Fréquent : Céphalée. Affections cardiaques : Fréquent : Tachycardie. Affections respiratoires, thoraciques et médiastinales : Fréquent : Rhinopharyngite. Affections gastro-intestinales : Très fréquent : Nausées, vomissements. Fréquent : Douleurs abdominales. Affections musculosquelettiques et systémiques : Fréquent : Douleurs des extrémités, arthralgies, douleurs musculosquelettiques. Troubles généraux et anomalies au site d'administration : Très fréquent : Fièvre, fatigue. Fréquent : Réaction aiguë, douleurs. Investigations : Très fréquent : Hypocalcémie. Fréquent : Hyphophosphatémie. - Les réactions indésirables apparaissant avec des fréquences < 5 % ont été médicalement évaluées et il a été montré que ces cas sont en accord avec le profil de sécurité d'emploi bien établi de Zometa (cf Effets indésirables).
- Chez l'enfant atteint d'ostéogenèse imparfaite sévère, l'acide zolédronique semble être associé à des risques plus prononcés de réaction aiguë, d'hypocalcémie ou de tachycardie inexpliquée, en comparaison au pamidronate, mais cette différence diminue après plusieurs perfusions.
- L'Agence européenne du médicament a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec Zometa dans tous les sous-groupes de la population pédiatrique dans le traitement de l'hypercalcémie induite par des tumeurs et dans la prévention des complications osseuses chez des patients atteints de pathologie maligne à un stade avancé avec atteinte osseuse (cf Posologie et Mode d'administration pour les informations concernant l'usage pédiatrique).
PHARMACOCINÉTIQUE |
Des perfusions uniques et multiples de 5 et 15 minutes de 2, 4, 8 et 16 mg d'acide zolédronique chez 64 patients ayant des métastases osseuses ont fourni les données pharmacocinétiques suivantes, qui sont dose-indépendantes.
Après le début de la perfusion d'acide zolédronique, les concentrations plasmatiques de la substance ont augmenté rapidement pour atteindre leurs pics à la fin de la perfusion et pour ensuite diminuer rapidement à moins de 10 % du pic après 4 heures et à moins de 1 % du pic après 24 heures, avec une période prolongée ultérieure de concentration très basse, ne dépassant pas 0,1 % du pic avant la seconde perfusion au jour 28.
L'acide zolédronique administré par voie intraveineuse a une élimination triphasique : une disparition rapide biphasique de la circulation sanguine, avec des demi-vies de t½ alpha 0,24 heure et t½ ß 1,87 heure, suivie par une longue phase d'élimination avec une demi-vie d'élimination terminale de t½ gamma 146 heures. Il n'y a pas d'accumulation plasmatique de la substance après administration de doses multiples tous les 28 jours.
L'acide zolédronique n'est pas métabolisé et est excrété sous forme inchangée par voie rénale. Au-delà des 24 premières heures, 39 % ± 16 de la dose administrée sont retrouvés dans les urines, alors que la quantité restante est principalement liée au tissu osseux. A partir du tissu osseux, il est libéré très lentement dans la circulation systémique et éliminé par voie rénale. La clairance corporelle totale est de 5,04 l/h ± 2,5, est indépendante de la dose et non affectée par le sexe, l'âge, la race et le poids corporel. L'augmentation de la durée de perfusion de 5 à 15 minutes a entraîné une réduction de 30 % de la concentration de l'acide zolédronique en fin de perfusion mais n'a pas modifié l'aire sous la courbe de la concentration plasmatique par rapport au temps.
Comme cela est observé avec les autres bisphosphonates, la variabilité entre les patients des paramètres pharmacocinétiques de l'acide zolédronique est élevée.
Aucune donnée pharmacocinétique concernant l'acide zolédronique n'est disponible chez les patients ayant une hypercalcémie ou chez les patients atteints d'une insuffisance hépatique.
In vitro, l'acide zolédronique n'inhibe pas les isoenzymes humaines du cytochrome P450, il ne subit pas de biotransformation, et dans les études animales, moins de 3 % de la dose administrée sont retrouvés dans les fèces, suggérant l'absence de rôle significatif de la fonction hépatique dans la pharmacocinétique de l'acide zolédronique.
La clairance rénale de l'acide zolédronique était corrélée à la clairance de la créatinine, la clairance rénale représentant 75 % ± 33 de la clairance de la créatinine, qui atteint une moyenne de 84 ml/min ± 29 (extrêmes : 22 et 143 ml/min) chez 64 patients atteints d'un cancer. L'analyse de la population a montré que pour les patients ayant une clairance de la créatinine de 20 ml/min (insuffisance rénale sévère) ou de 50 ml/min (insuffisance rénale modérée), la clairance prédictive correspondante de l'acide zolédronique devrait être respectivement de 37 % ou de 72 % de celle d'un patient ayant une clairance de la créatinine de 84 ml/min. Des données pharmacocinétiques encore limitées sont disponibles uniquement chez des patients avec une insuffisance rénale sévère (clairance de la créatinine < 30 ml/min).
L'acide zolédronique ne possède pas d'affinité pour les composants cellulaires sanguins et la liaison aux protéines plasmatiques est faible (approximativement 56 %) et indépendante de la concentration de l'acide zolédronique.
- Populations particulières :
- Enfants :
- Les données limitées de pharmacocinétique chez l'enfant atteint d'ostéogenèse imparfaite sévère suggèrent que la pharmacocinétique de l'acide zolédronique chez l'enfant âgé de 3 à 17 ans est identique à celle de l'adulte à des taux similaires en mg/kg. L'âge, le poids, le sexe et la clairance de la créatinine semblent ne pas avoir d'effet sur l'exposition à l'acide zolédronique.
SÉCURITE PRÉCLINIQUE |
- Toxicité aiguë :
- La dose non létale la plus élevée en administration intraveineuse unique était de 10 mg/kg de poids corporel chez la souris et de 0,6 mg/kg chez le rat.
- Toxicité subchronique et chronique :
- L'acide zolédronique a été bien toléré lorsqu'il a été administré par voie sous-cutanée chez le rat et par voie intraveineuse chez le chien, à des doses allant jusqu'à 0,02 mg/kg/j pendant 4 semaines. L'administration de 0,001 mg/kg/j par voie sous-cutanée chez le rat et de 0,005 mg/kg une fois tous les 2 à 3 jours par voie intraveineuse chez le chien sur une période allant jusqu'à 52 semaines a été également bien tolérée.
- Le résultat le plus fréquent dans les études à doses répétées est une augmentation de la spongiose primaire dans les métaphyses des os longs chez les animaux en cours de croissance à presque toutes les doses, un résultat qui reflète l'activité pharmacologique du produit sur la résorption osseuse.
- Les marges de sécurité relatives aux effets rénaux étaient étroites dans les études animales d'administration répétée par voie parentérale et à long terme, mais les taux cumulés sans effets indésirables observés (NOAEL) à dose unique (1,6 mg/kg) et à doses répétées jusqu'à un mois (0,06-0,6 mg/kg/j) n'ont pas montré d'effets rénaux à des doses équivalentes ou excédant la plus forte dose thérapeutique envisagée chez l'homme.
- L'administration répétée à plus long terme de doses d'acide zolédronique voisines de la plus forte dose thérapeutique envisageable chez l'homme a produit des effets toxiques sur d'autres organes incluant le tractus gastro-intestinal, le foie, la rate, les poumons, et au niveau du site d'injection IV.
- Toxicité sur la reproduction :
- L'acide zolédronique a présenté un effet tératogène chez le rat à des doses sous-cutanées >= 0,2 mg/kg. Bien qu'aucun effet tératogène ou foetotoxique n'ait été observé chez le lapin, une toxicité maternelle a été mise en évidence.
- Mutagénicité et pouvoir carcinogène :
- L'acide zolédronique ne s'est pas révélé mutagène au cours des tests de mutagénicité, et les études de carcinogénicité n'ont pas mis en évidence de pouvoir carcinogène. Une dystocie a été observée chez le rat à la plus faible dose testée (0,01 mg/kg de poids corporel)..
INCOMPATIBILITÉS |
Pour éviter les incompatibilités potentielles, la solution de Zometa doit être diluée dans une solution de chlorure de sodium à 0,9 % m/v ou une solution de glucose à 5 % m/v.
La solution à diluer de Zometa ne doit pas être mélangée avec des solutions contenant du calcium ou avec d'autres solutions pour perfusion contenant des cations divalents telle que la solution de Ringer lactate et doit être administrée par voie de perfusion séparée en solution intraveineuse unique.
Les études menées avec des flacons en verre, ainsi qu'avec plusieurs types de poches et de tubulures à perfusion en chlorure de polyvinyle, polyéthylène et polypropylène (préremplies avec une solution de chlorure de sodium à 0,9 % m/v ou une solution de glucose à 5 % m/v) n'ont montré aucune incompatibilité avec Zometa.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 3 ans.
Il n'y a pas de précautions particulières de conservation.
- Après dilution :
- Après dilution en conditions aseptiques, il est préférable d'utiliser immédiatement le produit dilué. Dans le cas où il ne serait pas utilisé immédiatement, la durée et les conditions de stockage avant l'utilisation sont sous la responsabilité de l'utilisateur. Le temps total entre la dilution, le stockage au réfrigérateur (entre 2 °C et 8 °C) et la fin de l'administration ne doit pas dépasser 24 heures.
- La solution de Zometa est chimiquement et physiquement stable pendant 24 heures entre 2 °C et 8 °C après dilution dans 100 ml de sérum physiologique ou de solution de glucose 5 % m/v.
MODALITÉS MANIPULATION/ÉLIMINATION |
Avant l'administration, les 5 ml de la solution concentrée ou le volume prélevé nécessaire de la solution concentrée doivent être dilués avec 100 ml de solution de perfusion exempte de calcium (solution de chlorure de sodium à 0,9 % m/v ou solution de glucose à 5 % m/v).
La solution doit être laissée à température ambiante avant administration si elle a été réfrigérée.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| Médicament soumis à une surveillance particulière pendant le traitement. | |
| AMM | EU/1/01/176/004 ; CIP 3400936187696 (RCP rév 25.01.2010). |
| Prix : | 314,76 euro(s) (1 flacon). |
| Remb Séc soc à 65 %. Collect. | |
| Titulaire de l'AMM : Novartis Europharm Limited, Wimblehurst Road, Horsham. West Sussex. RH12 5AB. Royaume-Uni. | |
Novartis Pharma SAS
2-4, rue Lionel-Terray. 92500 Rueil-Malmaison
Tél : 01 55 47 60 00
Information et Communication Médicales :
Tél : 01 55 47 66 00 E-mail : icm.phfr@novartis.com
Site web : http://www.novartis.fr
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale
