FIRMAGON®
dégarélix
FORMES et PRÉSENTATIONS |
Poudre (blanche à presque blanche) et solvant (solution limpide, incolore) pour solution injectable SC à 80 mg (traitement d'entretien) : Flacon de poudre + flacon de 6 ml de solvant + seringue de 5 ml (avec un double marquage volumétrique à 4,0 ml et 4,2 ml) + 2 adaptateurs-flacon + 1 aiguille pour l'injection (25 G 0,5 x 25 mm), boîte unitaire.
COMPOSITION |
| Poudre : | p flacon |
| Dégarélix (DCI) acétate exprimé en dégarélix | 120 mg |
| ou | 80 mg |
Solvant : eau ppi.
Après reconstitution, chaque ml de solution contient 40 mg de dégarélix (Firmagon 120 mg) ou 20 mg de dégarélix (Firmagon 80 mg).
INDICATIONS |
POSOLOGIE ET MODE D'ADMINISTRATION |
| Initiation du traitement | Traitement d'entretien - administration mensuelle |
| 240 mg administrés en 2 injections sous-cutanées de 120 mg chacune | 80 mg administrés en 1 injection sous-cutanée |
La première dose du traitement d'entretien doit être administrée 1 mois après la dose d'initiation du traitement.
La réponse thérapeutique au dégarélix est évaluée par l'examen clinique et les dosages sanguins de l'antigène spécifique de la prostate (PSA). Les études cliniques ont montré que l'inhibition de la sécrétion de testostérone (T) débute immédiatement après l'administration de la dose d'initiation du traitement. Un taux sérique de testostérone, correspondant à la castration médicale (T <= 0,5 ng/ml), est atteint trois jours après chez 96 % des patients et un mois après, chez 100 % des patients. Il a été montré qu'après 1 an de traitement à la dose d'entretien, la suppression de la sécrétion de testostérone (T <= 0,5 ng/ml) se maintenait chez 97 % des patients.
En l'absence de réponse clinique optimale, il faut s'assurer que le taux sérique de testostérone obtenu correspond bien à une suppression androgénique.
Le dégarélix n'induisant pas de pic de testostérone, il n'est pas nécessaire de prescrire un anti-androgène lors de l'instauration du traitement.
- Populations particulières :
-
- Patients âgés, insuffisants hépatiques ou insuffisants rénaux :
Il n'est pas nécessaire d'ajuster la posologie chez les patients âgés ou atteints d'insuffisance hépatique ou rénale légère ou modérée (cf Pharmacocinétique). En l'absence d'étude chez des patients souffrant d'une insuffisance hépatique ou rénale sévère, la prudence s'impose (cf Mises en garde et Précautions d'emploi).
- Firmagon n'est pas indiqué chez la femme, l'enfant et l'adolescent.
- Patients âgés, insuffisants hépatiques ou insuffisants rénaux :
Mode d'administration :
Avant l'administration, il faut procéder à la reconstitution de Firmagon. Pour les instructions de reconstitution et d'administration, cf Modalités de manipulation et d'élimination.
Firmagon doit être administré uniquement par voie sous-cutanée. Ne pas administrer par voie intraveineuse.
En l'absence d'étude, l'administration par voie intramusculaire n'est pas recommandée.
L'administration de Firmagon s'effectue par injection sous-cutanée dans la région abdominale. Comme pour tout médicament administré par voie sous-cutanée, le site d'injection doit être modifié périodiquement. Les injections doivent être réalisées dans une partie du corps non exposée à la pression, par exemple à distance de la taille, de la ceinture ou des côtes.
CONTRE-INDICATIONS |
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
- Effet sur l'intervalle QT/QTc :
- L'inhibition prolongée de la sécrétion androgénique est susceptible d'être à l'origine d'un allongement de l'intervalle QT. Dans l'étude pivot comparant Firmagon à la leuproréline, des électrocardiogrammes périodiques (mensuels) ont été réalisés : les deux traitements ont montré des intervalles QT/QTc dépassant 450 msec chez 20 % des patients et dépassant 500 msec chez respectivement 1 % et 2 % des patients ayant reçu du dégarélix ou de la leuproréline (cf Pharmacodynamie).
- Firmagon n'a pas été étudié chez les patients ayant eu des épisodes de QT corrigé supérieur à 450 msec, ayant des antécédents ou présentant des facteurs de risque de torsades de pointes, ou traités par un médicament susceptible de prolonger l'intervalle QT. Le rapport bénéfice/risque de Firmagon doit donc être évalué d'une façon approfondie chez ces patients (cf Interactions, Effets indésirables).
- Insuffisance hépatique :
- Les essais cliniques à long terme du dégarélix n'ont pas inclus de patients ayant ou suspectés d'avoir une insuffisance hépatique. Une augmentation faible et transitoire des taux des transaminases ALAT et ASAT a été observée, sans élévation du taux de bilirubine ni survenue d'une symptomatologie clinique. Chez les patients ayant ou suspectés d'avoir une insuffisance hépatique, une surveillance de la fonction hépatique est recommandée durant le traitement. La pharmacocinétique du dégarélix a été étudiée chez des patients présentant une insuffisance hépatique légère à modérée après administration d'une dose unique intraveineuse (cf Pharmacocinétique).
- Insuffisance rénale :
- Aucune étude n'a été réalisée chez les patients présentant une insuffisance rénale sévère. La prudence s'impose donc chez ces patients.
- Hypersensibilité :
- Aucune étude n'a été réalisée chez les patients ayant des antécédents d'asthme sévère non traité, de manifestations anaphylactiques ou d'urticaire sévère ou d'angio-oedème.
- Modification de la densité osseuse :
- Une diminution de la densité osseuse a été rapportée dans la littérature médicale chez des hommes ayant subi une orchidectomie ou ayant été traités par un agoniste de la GnRH. Des effets sur la densité osseuse sont prévisibles après inhibition prolongée de la sécrétion de testostérone. La densité osseuse n'a pas été mesurée lors du traitement par dégarélix.
- Tolérance au glucose :
- Une diminution de la tolérance au glucose a été observée chez des patients ayant subi une orchidectomie ou ayant été traités par un agoniste de la GnRH. La survenue ou l'aggravation d'un diabète est possible. Une surveillance plus fréquente de la glycémie peut donc être nécessaire chez les patients recevant un traitement de privation androgénique. L'effet du dégarélix sur les concentrations d'insuline ou de glucose n'a pas été étudié.
INTERACTIONS |
Compte tenu de la possible prolongation de l'intervalle QTc lors de la privation androgénique, l'utilisation concomitante de dégarélix avec des médicaments connus pour allonger l'intervalle QTc ou capables d'induire des torsades de pointes tels que les anti-arythmiques de la classe IA (par exemple quinidine, disopyramide) ou de la classe III (par exemple amiodarone, sotalol, dofétilide, ibutilide), méthadone, cisapride, moxifloxacine, antipsychotiques, etc., doit être évaluée avec précaution (cf Mises en garde et Précautions d'emploi).
Le dégarélix n'est pas un substrat pour le CYP450 humain. Il ne s'est avéré, in vitro, ni inducteur ni inhibiteur de CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, ni de CYP3A4/5 à un niveau significatif. Une interaction pharmacocinétique cliniquement significative avec d'autres médicaments métabolisés par ces systèmes enzymatiques est donc peu probable.
FERTILITÉ/GROSSESSE/ALLAITEMENT |
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
Les principaux événements indésirables au site d'injection ont été une douleur et un érythème, rapportés chez respectivement 28 % et 17 % des patients. La survenue d'un gonflement (6 %), d'une induration (4 %) ou d'un nodule (3 %) a été moins fréquemment rapportée. Ces événements sont survenus essentiellement à la phase d'initiation du traitement, alors que durant le traitement d'entretien (à la dose de 80 mg), l'incidence de ces événements a été, pour 100 injections, de 3 pour la douleur et < 1 pour la survenue d'un érythème, d'un gonflement, d'un nodule et d'une induration. Les événements rapportés ont été le plus souvent transitoires, d'intensité faible à modérée et n'ont conduit que très rarement à l'arrêt du traitement (< 1 %).
La fréquence des effets indésirables listés ci-dessous est définie selon la convention suivante : très fréquent (>= 1/10) ; fréquent (>= 1/100 à < 1/10) ; peu fréquent (>= 1/1000 à < 1/100). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
- Fréquence des effets indésirables rapportés chez 1259 patients traités sur un total de 1781 patients-années (études de phase II et III) :
- (Système MedDRA de Classification des Organes [SCO])
- Affections hématologiques et du système lymphatique :
- Fréquent : Anémie*.
- Fréquent : Anémie*.
- Affections du système immunitaire :
- Peu fréquent : Hypersensibilité.
- Peu fréquent : Hypersensibilité.
- Troubles du métabolisme et nutritionnel :
- Fréquent : Prise de poids*.
- Peu fréquent : Hyperglycémie/diabète sucré, augmentation du taux de cholestérol, perte de poids, perte d'appétit, modification de la calcémie.
- Fréquent : Prise de poids*.
- Affections psychiatriques :
- Fréquent : Insomnie.
- Peu fréquent : Dépression, baisse de libido*.
- Fréquent : Insomnie.
- Affections du système nerveux :
- Fréquent : Vertiges, céphalées.
- Peu fréquent : Ralentissement intellectuel, hypoesthésie.
- Fréquent : Vertiges, céphalées.
- Affections oculaires :
- Peu fréquent : Vision trouble.
- Peu fréquent : Vision trouble.
- Affections cardiaques :
- Peu fréquent : Arythmie cardiaque (y compris fibrillation auriculaire), palpitations, allongement du QT* (cf Mises en garde et Précautions d'emploi, Interactions).
- Peu fréquent : Arythmie cardiaque (y compris fibrillation auriculaire), palpitations, allongement du QT* (cf Mises en garde et Précautions d'emploi, Interactions).
- Affections vasculaires :
- Très fréquent : Bouffées de chaleur*.
- Peu fréquent : Hypertension, réaction vasovagale (y compris hypotension).
- Très fréquent : Bouffées de chaleur*.
- Affections respiratoires, thoraciques et médiastinales :
- Peu fréquent : Dyspnée.
- Peu fréquent : Dyspnée.
- Affections gastro-intestinales :
- Fréquent : Diarrhées, nausées.
- Peu fréquent : Constipation, vomissements, douleur abdominale, gêne abdominale, sécheresse de la bouche.
- Fréquent : Diarrhées, nausées.
- Affections hépatobiliaires :
- Fréquent : Augmentation des transaminases hépatiques.
- Peu fréquent : Augmentation de la bilirubine, augmentation de la phosphatase alcaline.
- Fréquent : Augmentation des transaminases hépatiques.
- Affections de la peau et du tissu sous-cutané :
- Fréquent : Hyperhidrose (dont sueurs nocturnes)*, rash.
- Peu fréquent : Urticaire, nodule cutané, alopécie, prurit, érythème.
- Fréquent : Hyperhidrose (dont sueurs nocturnes)*, rash.
- Affections musculosquelettiques et systémiques :
- Fréquent : Douleur et gêne musculosquelettiques.
- Peu fréquent : Ostéoporose/ostéopénie, arthralgie, faiblesse musculaire, spasmes musculaires, gonflement/raideur des articulations.
- Fréquent : Douleur et gêne musculosquelettiques.
- Affections du rein et des voies urinaires :
- Peu fréquent : Pollakiurie, mictions impérieuses, dysurie, nycturie, insuffisance rénale, incontinence.
- Peu fréquent : Pollakiurie, mictions impérieuses, dysurie, nycturie, insuffisance rénale, incontinence.
- Affections des organes de reproduction et du sein :
- Fréquent : Gynécomastie*, atrophie testiculaire*, dysfonction érectile*.
- Peu fréquent : Douleur testiculaire, douleur mammaire, douleur pelvienne, irritation génitale, trouble de l'éjaculation.
- Fréquent : Gynécomastie*, atrophie testiculaire*, dysfonction érectile*.
- Troubles généraux et anomalies au site d'administration :
- Très fréquent : Réaction au site d'injection.
- Fréquent : Frissons, fièvre, fatigue*, syndromes pseudo-grippaux.
- Peu fréquent : Malaise, oedème périphérique.
- Très fréquent : Réaction au site d'injection.
-
*
Conséquence physiologique connue de la suppression de la sécrétion de testostérone.
- Modifications des valeurs biologiques :
- Les modifications des valeurs biologiques observées au cours de l'année de traitement dans l'étude pivot de phase III (N = 409) ont été similaires avec les 2 produits : le dégarélix et un agoniste à la GnRH (leuproréline) utilisé comme comparateur. Des valeurs des transaminases hépatiques (ALAT, ASAT et GGT) nettement anormales (> 3 fois la limite supérieure de la normale du laboratoire) étaient observées chez 2-6 % des patients ayant des valeurs normales avant le traitement. Une diminution marquée de l'hématocrite (<= 0,37) et du taux d'hémoglobine (<= 115 g/l) a été observée chez respectivement 40 % et 13 à 15 % des patients ayant des valeurs normales avant le traitement. La part qui revient au cancer de la prostate ou à la suppression androgénique pour expliquer cette diminution n'est pas clairement définie. Chez les patients ayant des valeurs normales avant le traitement, des valeurs nettement anormales de kaliémie (>= 5,8 mmol/l), de créatininémie (>= 177 µmol/l) et d'urée sanguine (>= 10,7 mmol/l) ont été observées respectivement chez 6 %, 2 % et 15 % des patients traités par dégarélix et chez 3 %, 2 % et 14 % des patients traités par leuproréline.
- Modifications de l'électrocardiogramme :
- Les modifications observées à l'ECG au cours d'une année de traitement dans l'étude pivot de phase III (N = 409) ont été similaires avec les 2 produits : le dégarélix et un agoniste à la GnRH (leuproréline) utilisé comme comparateur. Un QTcF >= 500 msec a été observé chez trois (< 1 %) des 409 patients du groupe dégarélix et chez quatre (2 %) des 201 patients du groupe leuproréline 7,5 mg. La médiane de modification du QTcF tout au long de l'étude a été de 12,0 msec avec le dégarélix et de 16,7 msec avec la leuproréline.
SURDOSAGE |
PHARMACODYNAMIE |
Classe pharmacothérapeutique : autres antagonistes d'hormone et agents apparentés, code ATC : L02BX02.
Le dégarélix est un antagoniste sélectif de l'hormone entraînant la libération de gonadotrophines (GnRH). Il se fixe de façon compétitive et réversible sur les récepteurs GnRH de l'hypophyse, entraînant ainsi rapidement une réduction de la libération des gonadotrophines, hormone lutéinisante (LH) et hormone folliculostimulante (FSH), et donc de la sécrétion de testostérone (T) par les testicules. Le cancer de la prostate, androgénodépendant, répond au traitement qui inhibe la production d'androgène. Contrairement aux agonistes de la GnRH, les antagonistes de la GnRH n'induisent pas de pic de LH, responsable du pic de testostérone/d'une stimulation de la tumeur et d'une possible exacerbation des symptômes (effet « flare up »), lors de l'initiation du traitement.
Une dose unique de 240 mg de dégarélix, suivie d'une dose d'entretien mensuelle de 80 mg entraîne la diminution rapide des taux de LH, de FSH et par conséquent, de testostérone. La concentration plasmatique de dihydrotestostérone (DHT) diminue de manière similaire à celle de la testostérone.
Le dégarélix est efficace pour supprimer la sécrétion de testostérone et la maintenir sous le seuil équivalent à une castration médicale (0,5 ng/ml). Le traitement d'entretien à la dose mensuelle de 80 mg assure le maintien de la suppression de la sécrétion de testostérone pendant au moins un an chez 97 % des patients. La valeur médiane du taux de testostérone après un an de traitement était de 0,087 ng/ml (intervalle interquartile 0,06-0,15), n = 167.
- Résultats de l'étude pivot de phase III :
- L'efficacité et la tolérance du dégarélix ont été évaluées dans une étude ouverte, multicentrique, randomisée, comparative, avec groupes parallèles. L'objectif de l'étude était d'évaluer l'efficacité et la tolérance de deux doses différentes de dégarélix, administrées une fois par mois, par voie sous-cutanée ; une dose initiale (initiation du traitement) de 240 mg (40 mg/ml), suivie de doses mensuelles de 160 mg (40 mg/ml) ou de 80 mg (20 mg/ml), comparativement à un traitement par la leuproréline 7,5 mg par voie intramusculaire, chez des patients atteints d'un cancer de la prostate et requérant une thérapie de suppression androgénique. Au total 620 patients ont été randomisés dans l'un des 3 groupes de traitement et 504 des patients (81 %) ont terminé l'étude. 41 patients (20 %) du groupe dégarélix à la dose de 240/80 mg et 32 patients (16 %) du groupe leuproréline sont sortis prématurément de l'étude.
- Parmi les 610 patients traités :
- 31 % avaient un cancer de la prostate localisé ;
- 29 % avaient un cancer de la prostate localement avancé ;
- 20 % avaient un cancer de la prostate métastatique ;
- 7 % avaient un état métastatique inconnu ;
- 13 % avaient subi précédemment une chirurgie ou une radiothérapie à but curatif et présentaient une augmentation des PSA.
- 31 % avaient un cancer de la prostate localisé ;
- Les données démographiques de base de la population à l'inclusion étaient comparables dans chaque bras de l'étude. L'âge médian était 74 ans (extrêmes : 47-98). L'objectif principal de l'étude était de démontrer l'efficacité du dégarélix en termes d'obtention et de maintien de la suppression de la sécrétion de testostérone à un seuil inférieur à 0,5 ng/ml, pendant une durée de traitement de 12 mois.
- C'est la dose d'entretien de dégarélix la plus faible, 80 mg, qui a été retenue.
- Obtention de taux sériques de testostérone (T) <= 0,5 ng/ml :
- Firmagon est efficace pour obtenir une suppression rapide de la sécrétion de testostérone : cf tableau 1.
-
Tableau 1 : Pourcentage de patients ayant une testostéronémie T <= 0,5 ng/ml après l'instauration du traitement. Temps Dégarélix 240/80 mg en SC Leuproréline 7,5 mg en IM Jour 1 52 % 0 % Jour 3 96 % 0 % Jour 7 99 % 1 % Jour 14 100 % 18 % Jour 28 100 % 100 %
- Prévention des pics de testostérone :
- Le pic était défini par une augmentation >= 15 % du taux de testostérone, par rapport aux valeurs de base, au cours des 2 premières semaines.
- Aucun des patients traités par dégarélix n'a présenté de pic de testostérone. Une diminution moyenne de 94 % de la concentration en testostérone a été observée à J3. Chez la plupart des patients traités par leuproréline, des pics de testostéronémie sont survenus ; une augmentation moyenne de 65 % de la concentration en testostérone était observée à J3. La différence était statistiquement significative (p < 0,001).
-
Figure 1 : Évolution du taux de testostérone, exprimée en pourcentage, de J0 à J28 dans chaque groupe de traitement (valeur médiane avec intervalles interquartiles). 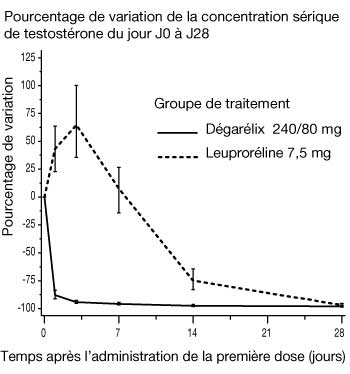
- Le critère principal de l'étude était le taux de suppression de la sécrétion de testostérone après un an de traitement par dégarélix ou leuproréline. Au cours de la phase initiale du traitement, le dégarélix n'a pas montré de bénéfice clinique comparé à celui de la leuproréline administrée en association avec un anti-androgène.
- Effet à long terme :
- Le succès en termes de réponse était défini comme l'obtention d'une castration médicale à J28 et son maintien sur une période de 364 jours, période pendant laquelle aucune concentration de testostérone ne devait être supérieure à 0,5 ng/ml.
-
Tableau 2 : Probabilité cumulée d'une concentration sérique de testostérone <= 0,5 ng/ml de J28 à J364. Dégarélix 240/80 mg n = 207 Leuproréline 7,5 mg n = 201 Nombre de répondeurs 202 194 Taux de réponse 97,2 % 96,4 % (Intervalles de confiance)* (93,5 ; 98,8 %) (92,5 ; 98,2 %) -
*
Valeurs estimées par Kaplan Meier dans chaque groupe.
- Baisse de l'antigène spécifique de la prostate (PSA) :
- La taille de la tumeur n'a pas été mesurée directement au cours de l'étude clinique mais la réduction de 95 % de la valeur médiane des PSA après 12 mois de traitement par dégarélix témoigne indirectement d'une réponse bénéfique sur la tumeur.
- Dans l'étude, la valeur médiane de base du PSA était :
- pour le groupe dégarélix (240/80 mg) : 19,8 ng/ml (intervalle interquartile : P25 9,4 ng/ml, P75 46,4 ng/ml) ;
- pour le groupe leuproréline 7,5 mg : 17,4 ng/ml (intervalle interquartile : P25 8,4 ng/ml, P75 56,5 ng/ml).
- pour le groupe dégarélix (240/80 mg) : 19,8 ng/ml (intervalle interquartile : P25 9,4 ng/ml, P75 46,4 ng/ml) ;
-
Figure 2 : Évolution du taux de PSA, exprimée en pourcentage, de J0 à J56, dans chaque groupe de traitement (valeur médiane avec intervalles interquartiles) 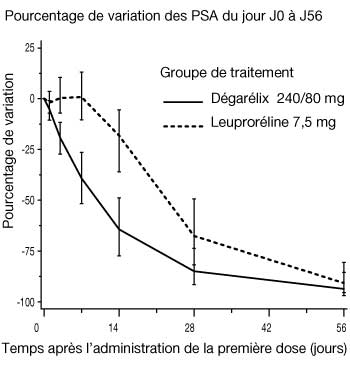
- Aux jours prédéterminés d'analyse, J14 et J28, cette différence était statistiquement significative (p < 0,001).
- Le taux d'antigènes spécifiques de la prostate (PSA) avait diminué de 64 %, 2 semaines après l'administration de dégarélix, de 85 % après 1 mois, de 95 % après 3 mois, et la suppression des PSA (environ 97 %) s'est maintenue pendant toute la durée (1 an) du traitement.
- De J56 à J364, il n'y avait pas de différence significative en pourcentage d'évolution par rapport aux valeurs initiales, entre le dégarélix et le comparateur.
- Dans l'étude pivot comparant Firmagon à la leuproréline, des ECG ont été réalisés périodiquement. Des intervalles QT/QTc supérieurs à 450 msec ont été observés chez environ 20 % des patients avec les deux traitements. Entre le début et la fin de l'étude, l'allongement médian a été de 12,0 msec avec Firmagon et de 16,7 msec avec la leuproréline.
- L'apparition d'anticorps anti-dégarélix a été observée chez 10 % des patients au cours du traitement par Firmagon d'une durée d'un an. Rien n'indique que l'efficacité ou la sécurité du traitement par Firmagon puisse être affectée par la formation d'anticorps après un an de traitement. On ne dispose pas de données d'efficacité et de sécurité relatives au développement d'anticorps au-delà d'un an.
PHARMACOCINÉTIQUE |
- Absorption :
- Dans l'étude pivot CS21, après l'administration sous-cutanée, à des patients souffrant d'un cancer de la prostate, de 240 mg de dégarélix, à la concentration de 40 mg/ml, l'AUC0-28 jours était de 635 (602-668) jour x ng/ml, la Cmax de 66,0 (61,0-71,0) ng/ml et la tmax de 40 (37-42) heures. Les valeurs résiduelles moyennes étaient d'environ 11-12 ng/ml après l'initiation du traitement et 11-16 ng/ml avec le traitement d'entretien à la dose de 80 mg, à la concentration de 20 mg/ml. Le dégarélix est éliminé de façon biphasique, avec une demi-vie terminale (t½) médiane d'environ 43 jours pour la dose administrée lors de l'initiation du traitement, et d'environ 28 jours pour la dose d'entretien, estimation basée sur une modélisation pharmacocinétique. La longue demi-vie après administration sous-cutanée est une conséquence de la très lente libération de dégarélix à partir du dépôt Firmagon formé au(x) point(s) d'injection. Le comportement pharmacocinétique du médicament est fortement influencé par sa concentration dans la solution injectable. Ainsi, la Cmax et la biodisponibilité tendent à diminuer avec l'augmentation de la concentration de la dose alors que la demi-vie augmente. Il en résulte que seule la dose à la concentration recommandée doit être utilisée.
- Distribution :
- Le volume de distribution chez le volontaire sain âgé est approximativement de 1 l/kg. La liaison aux protéines plasmatiques est estimée à environ 90 %.
- Métabolisme :
- Le dégarélix fait l'objet d'une dégradation commune aux peptides durant son passage dans le système hépatobiliaire. Il est principalement excrété dans les fèces sous forme de fragments peptidiques. Aucun métabolite significatif n'a été détecté dans les prélèvements sériques après administration sous-cutanée. Les études in vitro ont montré que le dégarélix n'est pas un substrat pour le CYP450 humain.
- Excrétion :
- Chez le volontaire sain, après une injection unique intraveineuse de dégarélix, environ 20 à 30 % sont excrétés dans les urines, ce qui suggère que 70 à 80 % sont excrétés via le système hépatobiliaire. Chez le volontaire sain âgé, la clairance du dégarélix après injection unique intraveineuse (0,864-49,4 µg/kg) est de 35-50 ml/h/kg.
- Populations particulières :
-
- Patients souffrant d'insuffisance rénale :
Aucune étude pharmacocinétique n'a été réalisée chez des patients atteints d'insuffisance rénale. Seulement, 20 % à 30 % environ de la dose administrée de dégarélix sont excrétés par le rein, sous forme inchangée. Une analyse des paramètres pharmacocinétiques à partir des données issues de l'étude pivot de phase III a montré que, chez les patients souffrant d'une insuffisance rénale légère à modérée, la clairance du dégarélix est réduite d'environ 23 % ; en conséquence, il n'est pas recommandé d'ajustement de la dose chez les patients souffrant d'une insuffisance rénale légère ou modérée. Pour les patients souffrant d'insuffisance rénale sévère, les données sont insuffisantes, et donc la prudence s'impose chez ce type de patients.
- Patients souffrant d'insuffisance hépatique :
La pharmacocinétique du dégarélix a été étudiée chez des patients atteints d'insuffisance hépatique légère à modérée. Par comparaison avec les sujets sains, aucun signe d'augmentation de l'exposition n'a été observé. L'ajustement de la dose n'est pas nécessaire chez les patients souffrant d'insuffisance hépatique légère ou modérée. Aucune étude n'a été réalisée chez les patients atteints de troubles hépatiques sévères. La prudence s'impose donc chez ce type de patients.
- Patients souffrant d'insuffisance rénale :
SÉCURITE PRÉCLINIQUE |
Les études de reproduction chez l'animal ont montré que le dégarélix entraînait une infertilité chez les mâles. Cet effet, dû à l'action pharmacologique du produit, était réversible.
Chez la femelle, les études de toxicité sur la reproduction réalisées avec le dégarélix ont donné les résultats attendus, du fait des propriétés pharmacologiques du produit. Le dégarélix a entraîné une augmentation, dose dépendante, du délai jusqu'à l'accouplement et la grossesse, un nombre réduit de corps jaune, ainsi qu'une augmentation du nombre d'échecs en phase pré et postimplantatoire, d'avortements, de morts précoces d'embryons/foetus, d'accouchements prématurés et une augmentation de la durée du travail.
Les études précliniques de sécurité, de pharmacologie de toxicité chronique, de génotoxicité et de pouvoir carcinogène, n'ont pas mis en évidence de risque particulier pour l'Homme. Les études in vitro et in vivo n'ont pas montré d'éléments en faveur d'un allongement du QT.
Chez le rat et le singe, les études de toxicité aiguë et chronique réalisées après administration sous-cutanée de dégarélix n'ont pas mis en évidence d'organe-cible. Chez l'animal, une irritation locale liée au produit, est apparue après l'administration par voie sous-cutanée de dégarélix à fortes doses.
INCOMPATIBILITÉS |
En l'absence d'étude de compatibilité, ce médicament ne doit pas être mélangé avec d'autres produits.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 3 ans.
Ce médicament n'exige pas de conditions de conservation particulières.
- Après reconstitution :
- La stabilité chimique et physique de la solution reconstituée, prête à l'emploi, a été montrée pendant 2 heures à 25 °C. D'un point de vue microbiologique, sauf si le mode de reconstitution élimine tous risques de contamination microbienne, le produit doit être injecté immédiatement. En cas de non-utilisation immédiate, l'utilisateur est seul responsable de la durée et des conditions de conservation de la solution reconstituée, prête à l'emploi.
MODALITÉS MANIPULATION/ÉLIMINATION |
Pas de recommandations spéciales pour l'élimination.
- Instructions d'utilisation :
- Les instructions de reconstitution doivent être soigneusement respectées.
- L'administration d'autres concentrations n'est pas recommandée car la formation du gel dépôt est influencée par la concentration. La solution reconstituée doit être limpide, sans particule visible.
- Note : les flacons ne doivent pas être secoués.
- La boîte de Firmagon 120 mg contient 2 sets de poudre et solvant qu'il faut préparer pour l'injection sous-cutanée (SC) : ainsi, chacune des instructions suivantes doit être reproduite une seconde fois.
- La boîte de Firmagon 80 mg contient 1 set de poudre et solvant qu'il faut préparer pour l'injection sous-cutanée (SC).
-
- Retirer l'emballage de protection des adaptateurs-flacon. Fixer les adaptateurs sur chacun des deux flacons de solvant et de poudre en exerçant une pression jusqu'à ce que le perforateur de l'adaptateur perce le bouchon du flacon et jusqu'à entendre le déclic de mise en place de l'adaptateur.
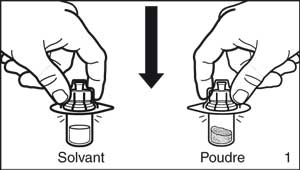
- Retirer l'emballage de protection de la seringue. Fixer la seringue au flacon de solvant en la vissant sur l'adaptateur.
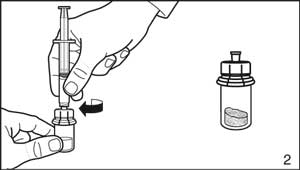
- Renverser le flacon la tête en bas et prélever 3,0 ml (Firmagon 120 mg) ou 4,2 ml (Firmagon 80 mg) de solvant dans la seringue.
Faites très attention de toujours prélever exactement le volume précisé car la quantité de solvant a une influence sur la reconstitution.
Retirer la seringue de l'adaptateur et jeter le flacon contenant le reste de solvant.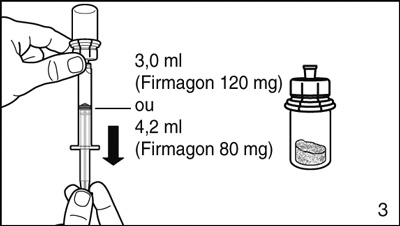
- Fixer la seringue au flacon de poudre en la vissant sur l'adaptateur. Transférer le solvant dans le flacon de poudre. Avec la seringue encore fixée à l'adaptateur, effectuer en douceur des rotations jusqu'à ce que le liquide paraisse limpide sans poudre ou particule visible. Dans le cas où la poudre adhérerait au flacon au-dessus de la surface du liquide, le flacon peut être légèrement incliné. Éviter de secouer pour prévenir la formation de mousse.
Un anneau de petites bulles d'air à la surface du liquide est acceptable. La procédure de reconstitution peut, dans certains cas, prendre jusqu'à 15 minutes, mais habituellement elle prend quelques minutes.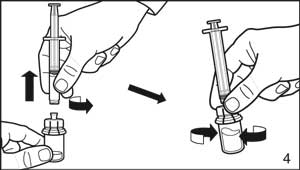
- Renverser le flacon la tête en bas, en le tenant en position verticale. Prélever 3,0 ml (Firmagon 120 mg) ou 4,0 ml (Firmagon 80 mg) de solution dans la seringue pour l'injection.
Faire très attention de toujours prélever exactement le volume précisé. Il peut être nécessaire d'incliner légèrement le flacon.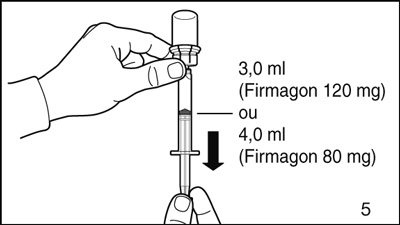
- Retirer la seringue de l'adaptateur-flacon et fixer l'aiguille destinée à l'injection sous-cutanée profonde sur la seringue. Éliminer, avec précaution, les bulles d'air.
- Pincer la peau de l'abdomen, et tirer vers le haut le tissu sous-cutané. Réaliser une injection sous-cutanée profonde. Pour cela, insérer profondément l'aiguille sous un angle d'au moins 45°.
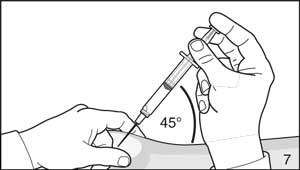
- Injecter 3,0 ml (Firmagon 120 mg) ou 4,0 ml (Firmagon 80 mg) immédiatement après reconstitution*.
- Ne pas injecter directement dans une veine. Remonter doucement le piston pour vérifier si du sang est aspiré. Si du sang apparaît dans la seringue, le produit ne peut plus être utilisé. Stopper la manipulation et jeter la seringue et l'aiguille. (Procéder à la reconstitution d'une nouvelle dose pour le patient).
- Firmagon 120 mg : Répéter la procédure de reconstitution pour une seconde dose. Choisir un point d'injection différent et injecter 3,0 ml.
- Retirer l'emballage de protection des adaptateurs-flacon. Fixer les adaptateurs sur chacun des deux flacons de solvant et de poudre en exerçant une pression jusqu'à ce que le perforateur de l'adaptateur perce le bouchon du flacon et jusqu'à entendre le déclic de mise en place de l'adaptateur.
- Attention : Ne pas faire d'injection dans les parties du corps exposées à une pression, par exemple à proximité de la ceinture, de la taille ou près des côtes.
-
*
La stabilité chimique et physique de la solution reconstituée, prête à l'emploi, a été montrée pendant 2 heures à 25 °C. D'un point de vue microbiologique, sauf si le mode de reconstitution élimine tout risque de contamination microbienne, le produit doit être injecté immédiatement. En cas de non-utilisation immédiate, l'utilisateur est seul responsable de la durée et des conditions de conservation de la solution reconstituée, prête à l'emploi.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| AMM | EU/1/08/504/002 ; CIP 3400939432748 (2009, RCP rév 14.10.2010) 120 mg. |
| EU/1/08/504/001 ; CIP 3400939432687 (2009, RCP rév 14.10.2010) 80 mg. |
| Prix : | 261.17 euros (120 mg). |
| 149.01 euros (80 mg). | |
| Remb Séc soc à 100 %. Collect. | |
Titulaire de l'AMM : Ferring Pharmaceuticals A/S.
Fabricant : FERRING GmbH-Kiel-Allemagne.
FERRING SAS
7, rue Jean-Baptiste-Clément. 94250 Gentilly
Tél (prix appel local) : 08 11 11 19 50
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale
