Comprimé pelliculé à 80 mg (en forme de gélule avec la gravure « 80 » sur une face ; jaune pâle à jaune) et à 120 mg (en forme de gélule avec la gravure « 120 » sur une face ; jaune pâle à jaune) : Boîtes de 28, sous plaquettes thermoformées transparentes de 14.
COMPOSITION
p cp
Fébuxostat (DCI)
80 mg
ou
120 mg
Excipients (communs) :
Noyau : lactose monohydraté, cellulose microcristalline, stéarate de magnésium, hyprolose, croscarmellose sodique, silice colloïdale hydratée. Pelliculage : Opadry II, jaune 85F42129 (polyvinylalcool, dioxyde de titane [E 171], macrogol 3350, talc, oxyde de fer jaune [E 172]).
Traitement de l'hyperuricémie chronique dans les cas où un dépôt d'urate s'est déjà produit (incluant des antécédents ou la présence de tophus et/ou d'arthrite goutteuse).
POSOLOGIE ET MODE D'ADMINISTRATION
La dose recommandée d'Adenuric est de 80 mg une fois par jour, administrée par voie orale, pendant ou en dehors des repas. Si l'uricémie est > 6 mg/dl (357 µmol/l) après 2 à 4 semaines de traitement, l'administration d'Adenuric 120 mg une fois par jour peut être envisagée. L'action d'Adenuric est suffisamment rapide pour permettre un nouveau dosage de l'uricémie après 2 semaines de traitement. L'objectif thérapeutique est la diminution et le maintien de l'uricémie au-dessous de 6 mg/dl (357 µmol/l). Un traitement préventif des crises de goutte est recommandé pendant au moins 6 mois (cf Mises en garde et Précautions d'emploi).
Populations particulières :
Insuffisance rénale :
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère à modérée. L'efficacité et la tolérance n'ont pas été totalement évaluées chez les patients présentant une insuffisance rénale sévère (clairance de la créatinine < 30 ml/min) : cf Pharmacocinétique.
Insuffisance hépatique :
La dose recommandée est de 80 mg chez les patients présentant une insuffisance hépatique légère. L'expérience clinique est limitée chez les patients présentant une insuffisance hépatique modérée. L'efficacité et la tolérance du fébuxostat n'ont pas été étudiées chez les patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh).
Sujet âgé :
Aucune adaptation posologique n'est nécessaire chez les patients âgés (cf Pharmacocinétique).
Enfant et adolescent :
En l'absence d'expérience clinique chez l'enfant et l'adolescent, l'utilisation de fébuxostat n'est pas recommandée chez ces patients.
Greffe d'organe :
En l'absence d'expérience clinique chez le patient ayant reçu une greffe d'organe, l'utilisation de fébuxostat n'est pas recommandée chez ces patients (cf Pharmacodynamie).
CONTRE-INDICATIONS
Hypersensibilité à la substance active ou à l'un des excipients (cf Effets indésirables).
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI
Affections cardiovasculaires :
Le traitement par fébuxostat n'est pas recommandé chez les patients atteints de cardiopathie ischémique ou d'insuffisance cardiaque congestive (cf Effets indésirables).
Crise de goutte :
Le traitement par fébuxostat ne doit pas être instauré avant la disparition complète d'une crise de goutte. Comme avec les autres hypo-uricémiants, des crises de goutte peuvent survenir en début de traitement en raison d'une variation de l'uricémie qui entraîne une mobilisation des cristaux d'urate à partir des dépôts tissulaires (cf Effets indésirables, Pharmacodynamie). Lors de l'instauration d'un traitement par fébuxostat, un traitement préventif de la crise de goutte par un anti-inflammatoire non stéroïdien ou par la colchicine est recommandé pendant au moins 6 mois (cf Posologie et Mode d'administration).
En cas de survenue d'une crise de goutte au cours du traitement, ne pas interrompre la prise de fébuxostat. Un traitement de la crise de goutte adapté à chaque patient doit être administré simultanément. La fréquence et l'intensité des crises de goutte diminuent lors de la poursuite du traitement par fébuxostat.
Dépôt de xanthine :
Comme avec les autres hypo-uricémiants, chez les patients ayant une production d'urate fortement accrue (par exemple affection maligne traitée, syndrome de Lesch-Nyhan), la concentration absolue de xanthine au niveau urinaire peut, dans de rares cas, augmenter suffisamment pour entraîner un dépôt dans les voies urinaires. En l'absence d'expérience clinique avec le fébuxostat dans cette population, son administration n'est pas recommandée chez ces patients.
Mercaptopurine/azathioprine :
L'administration du fébuxostat n'est pas recommandée chez les patients traités par mercaptopurine/azathioprine (cf Interactions).
Théophylline :
Le fébuxostat doit être prescrit avec prudence chez les patients traités par théophylline, et la théophyllinémie doit être surveillée au début du traitement par le fébuxostat (cf Interactions).
Affections hépatiques :
Les résultats combinés des études cliniques de phase III ont montré de légères anomalies du bilan hépatique chez des patients (5,0 %) traités par fébuxostat. La réalisation d'un bilan hépatique est recommandée avant l'instauration du traitement par fébuxostat et périodiquement par la suite, en fonction du jugement clinique (cf Pharmacodynamie).
Affections de la thyroïde :
Au cours des études d'extension en ouvert à long terme, une augmentation du taux de TSH (> 5,5 µUI/ml) a été observée chez des patients traités au long cours par fébuxostat (5,5 %). Le fébuxostat doit être prescrit avec prudence chez les patients présentant une altération de la fonction thyroïdienne (cf Pharmacodynamie).
Lactose :
Les comprimés de fébuxostat contiennent du lactose. Les patients présentant des troubles héréditaires rares d'intolérance au galactose, de déficit en lactase ou de malabsorption du glucose/galactose ne doivent pas prendre ce médicament.
INTERACTIONS
Interactions médicamenteuses :
Mercaptopurine/azathioprine :
Aucune étude d'interaction n'a été menée avec le fébuxostat, mais l'on sait que l'inhibition de la xanthine oxydase (XO) conduit à une augmentation des concentrations de mercaptopurine et d'azathioprine. En raison de son mécanisme d'action inhibiteur de la XO, l'administration concomitante de fébuxostat n'est pas recommandée.
Aucune étude d'interaction entre le fébuxostat et une chimiothérapie cytotoxique n'a été menée.
Aucune donnée n'est disponible quant à la sécurité d'emploi du fébuxostat au cours d'un traitement cytotoxique.
Théophylline :
Aucune étude d'interaction n'a été menée avec le fébuxostat, mais l'inhibition de la XO peut induire une élévation de la théophyllinémie (une inhibition du métabolisme de la théophylline a été décrite avec d'autres inhibiteurs de la XO). Il est recommandé d'être prudent en cas d'administration concomitante de ces deux principes actifs, et de surveiller la théophyllinémie en début de traitement par fébuxostat.
Naproxène et autres inhibiteurs de la glycuronidation :
Le métabolisme du fébuxostat dépend des enzymes UGT. Les médicaments qui inhibent la glycuronidation, tels les anti-inflammatoires non stéroïdiens et le probénécide, pourraient théoriquement affecter l'élimination du fébuxostat. Chez des volontaires sains, l'administration concomitante de fébuxostat et de naproxène 250 mg deux fois par jour a été associée à une augmentation de l'exposition au fébuxostat (Cmax 28 %, ASC 41 % et t½ 26 %). Au cours des études cliniques, l'administration de naproxène ou d'autres anti-inflammatoires non stéroïdiens ou inhibiteurs de la Cox 2 n'a pas été associée à une augmentation cliniquement significative des événements indésirables.
Le fébuxostat peut être administré de façon concomitante avec le naproxène sans qu'une adaptation de la posologie du fébuxostat ou du naproxène ne soit nécessaire.
Inducteurs de la glycuronidation :
Les inducteurs puissants des enzymes UGT peuvent accroître le métabolisme et diminuer l'efficacité du fébuxostat. Un contrôle de l'uricémie est donc recommandé une à deux semaines après le début d'un traitement par un inducteur puissant de la glycuronidation. A l'inverse, l'arrêt du traitement par un inducteur pourrait se traduire par une augmentation de la concentration plasmatique du fébuxostat.
Le fébuxostat peut être administré de façon concomitante avec la colchicine ou l'indométacine sans adaptation de la dose de l'une ou l'autre des substances actives.
Aucune adaptation posologique du fébuxostat n'est nécessaire en cas d'administration concomitante d'hydrochlorothiazide.
Aucune adaptation posologique de la warfarine n'est nécessaire en cas d'administration concomitante avec le fébuxostat. L'administration concomitante de fébuxostat (80 mg ou 120 mg en une prise par jour) et de warfarine n'a pas montré d'effet sur la pharmacocinétique de la warfarine chez des sujets sains. L'INR et l'activité du facteur VII n'ont pas non plus été affectés par la coadministration de fébuxostat.
Désipramine/substrats du CYP2D6 :
Le fébuxostat exerce un léger effet inhibiteur du CYP2D6 in vitro. Lors d'une étude chez le volontaire sain, l'administration de 120 mg d'Adenuric une fois par jour a conduit à une augmentation moyenne de 22 % de l'ASC de la désipramine, substrat du CYP2D6, témoignant d'un faible effet inhibiteur potentiel du fébuxostat sur le CYP2D6 in vivo. L'administration concomitante de fébuxostat avec d'autres substrats du CYP2D6 ne devrait donc pas nécessiter d'adaptation de la posologie de ces produits.
Antiacides :
La prise concomitante d'un antiacide contenant des hydroxydes de magnésium et d'aluminium a retardé l'absorption du fébuxostat (d'environ une heure) et a induit une diminution de 32 % de la Cmax, mais sans modification significative de l'ASC. Le fébuxostat peut donc être administré sans tenir compte de la prise concomitante d'un antiacide.
FERTILITÉ/GROSSESSE/ALLAITEMENT
Grossesse :
Les données recueillies sur un nombre très limité de grossesses n'ont pas révélé d'effet délétère du fébuxostat sur la grossesse ou sur le foetus/nouveau-né. Les études menées chez l'animal n'ont pas montré d'effets délétères directs ou indirects sur la gestation, le développement embryonnaire ou foetal ou la mise bas (cf Sécurité préclinique). Le risque potentiel en clinique n'est pas connu. Le fébuxostat ne doit pas être utilisé au cours de la grossesse.
Allaitement :
L'excrétion du fébuxostat dans le lait maternel n'est pas connue. Des études menées chez l'animal ont montré une excrétion du principe actif dans le lait et une altération du développement des petits allaités. Un risque pour le nourrisson allaité ne peut être exclu. Le fébuxostat ne doit pas être utilisé chez la femme qui allaite.
CONDUITE et UTILISATION DE MACHINES
Les effets du fébuxostat sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés. Comme avec les autres inhibiteurs de la xanthine oxydase, des réactions indésirables, telles que somnolence, sensations vertigineuses et paresthésies, ont été rapportées. Les patients doivent être prudents avant de conduire des véhicules, d'utiliser des machines ou de participer à des activités dangereuses tant qu'ils ne sont pas raisonnablement certains qu'Adenuric ne nuit pas à leurs performances.
EFFETS INDÉSIRABLES
Au total, 4072 patients ont reçu au moins une dose d'Adenuric (10 mg - 300 mg) au cours des études cliniques.
Résultats combinés des études randomisées et contrôlées de phase III :
Au cours des études randomisées et contrôlées de phase III, plus de 2500 patients ont été traités avec des doses comprises entre 40 mg et 120 mg : 1513 sujets inclus dans une étude de 26 semaines (CONFIRMS), 536 sujets inclus dans une étude de 28 semaines (APEX) et 507 sujets inclus dans une étude de 52 semaines (FACT). Les événements indésirables liés au traitement ont généralement été de sévérité légère ou modérée.
Les événements indésirables liés au traitement (selon le jugement de l'investigateur) le plus fréquemment rapportés ont été des anomalies du bilan hépatique (5,0 %), des diarrhées (2,7 %), des nausées (1,3 %), des céphalées (1,2 %) et des éruptions (1,2 %).
Des crises de goutte ont également été fréquemment observées juste après l'instauration du traitement et durant les premiers mois. Par la suite, la fréquence des crises de goutte décroît avec le temps.
Comme pour les autres hypo-uricémiants, une prophylaxie des crises de goutte est recommandée (cf Posologie et Mode d'administration, Mises en garde et Précautions d'emploi).
Comparativement au groupe allopurinol, il a été observé dans le groupe fébuxostat une incidence numériquement plus élevée des événements cardiovasculaires APTC (critères définis selon l'Anti-Platelet Trialists' Collaboration [APTC] rapportés par les investigateurs comprenant les décès pour cause cardiovasculaire, les infarctus du myocarde non fatals, les AVC non fatals) au cours des études APEX et FACT (1,3 événements par 100 patients-années contre 0,3) mais pas dans l'étude CONFIRMS. Les résultats combinés des études de phase III (études APEX, FACT et CONFIRMS) ont montré une incidence des évènements APTC rapportés par les investigateurs de 0,7 événement pour 100 patients-années contre 0,6 dans le groupe allopurinol. Au cours des études d'extension à long terme l'incidence des évènements APTC rapportés par les investigateurs était de 1,2 pour 100 patients-années dans le groupe fébuxostat contre 0,6 dans le groupe allopurinol. Aucune différence statistiquement significative n'a été observée et aucune relation de cause à effet n'a été établie avec le fébuxostat. Chez ces patients, les facteurs de risque identifiés ont été des antécédents d'athérosclérose et/ou d'infarctus du myocarde ou d'insuffisance cardiaque congestive.
Les événements indésirables fréquents (>= 1/100 à < 1/10), peu fréquents (>= 1/1000 à < 1/100) et rares (>= 1/10 000 à < 1/1000), considérés comme pouvant être liés au traitement (selon le jugement de l'investigateur), survenus dans les groupes traités par une dose comprise entre 40 mg et 120 mg et rapportés à plus d'une occasion dans la population totale traitée par fébuxostat sont mentionnés ci-dessous.
Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre de sévérité décroissante.
Tableau 1 : Événements indésirables liés au traitement lors des études randomisées et contrôlées de phase III :
Affections hématologiques et du système lymphatique :
Rare
Pancytopénie
Troubles du métabolisme et de la nutrition :
Fréquent*
Crises de goutte
Peu fréquent
Diminution de l'appétit
Rare
Prise/perte de poids, augmentation de l'appétit, anorexie, hyperlipidémie, crises de goutte
Affections psychiatriques :
Peu fréquent
Diminution de la libido, insomnie
Rare
Nervosité
Affections du système nerveux :
Fréquent
Céphalées
Peu fréquent
Sensations vertigineuses, paresthésies, somnolence, altération du goût, hypoesthésie
Affections auditives et du labyrinthe :
Rare
Acouphènes
Affections cardiaques :
Peu fréquent
Fibrillation auriculaire, palpitations, anomalies de l'ECG
Affections vasculaires :
Peu fréquent
Hypertension, bouffées vasomotrices
Affections respiratoires :
Peu fréquent
Dyspnée, infections des voies respiratoires supérieures
Affections du système de reproduction et des seins :
Rare
Dysfonction érectile
Troubles généraux et anomalies au site d'administration :
Peu fréquent
Fatigue, oedème, douleurs thoraciques, gêne dans la poitrine,
Rare
Soif
Modifications des paramètres biologiques :
Peu fréquent
Augmentation de l'amylasémie, diminution de la numération plaquettaire, augmentation de la créatininémie, diminution de l'hémoglobinémie, augmentation de l'urémie, augmentation de la triglycéridémie, augmentation de la cholestérolémie, diminution de l'hématocrite, augmentation de la lactate déshydrogénase dans le sang
Rare
Augmentation de la glycémie, allongement du temps de céphaline activée, diminution des globules rouges, augmentation des phosphatases alcalines dans le sang
*cf Pharmacodynamie pour l'incidence des crises de goutte dans les études de phase III randomisées et contrôlées.
**
Les résultats combinés des études de phase III ont montré des diarrhées non infectieuses et des anomalies de la fonction hépatique plus fréquentes chez les patients traités de façon concomitante par la colchicine.
***
Aucune éruption grave ni réaction sévère d'hypersensibilité n'a été observée lors des études cliniques.
Études d'extension en ouvert à long terme :
Au cours des études d'extension en ouvert à long terme (1143 patients), le nombre de patients traités par fébuxostat 40/80/120 mg était de 909 à 1 an, 781 à 2 ans, 348 à 3 ans et 60 à 4 et 5 ans. Les événements indésirables liés au traitement survenus lors de ces extensions d'études à long terme ont été similaires à ceux observés au cours des études de phase III (voir tableau 1). Les plus fréquents (selon le jugement de l'investigateur) ont été des cas d'anomalies du bilan hépatique, de diarrhées, de céphalées, d'éruption, d'hypertension et d'oedème.
Les événements indésirables suivants liés au traitement ont été rapportés plus d'une fois dans la population totale traitée par fébuxostat et ont été décrits comme peu fréquents chez les patients recevant le fébuxostat 40/80/120 mg lors des études d'extension à long terme (jusqu'à 5 ans, exposition > 2660 patients-années). Les événements indésirables liés au traitement qui n'ont pas été rapportés ou ont été rapportés à une fréquence plus faible à ces doses dans les résultats combinés des études de phase III sont les suivants : distension abdominale, lithiase biliaire, bronchite, prise de poids, augmentation de la créatininémie, diabète, hyperlipémie, arthrite, raideur musculaire, hyposmie, hémiparésie, toux, décoloration cutanée, lésion cutanée, bursite, protéinurie, pétéchie, dysfonction érectile, augmentation de la kaliémie, augmentation du taux sanguin de TSH, diminution de la numération lymphocytaire, diminution de la numération leucocytaire.
SURDOSAGE
Aucun cas de surdosage n'a été rapporté. Le traitement d'un surdosage doit être symptomatique et comporter des mesures de soutien.
PHARMACODYNAMIE
Groupe pharmacothérapeutique : inhibiteurs de la synthèse d'acide urique (code ATC : M04AA03).
Mécanisme d'action :
L'acide urique est le produit final du métabolisme des purines chez l'homme et résulte de la cascade hypoxanthine => xanthine => acide urique. Ces deux étapes sont catalysées par la xanthine oxydase (XO). Le fébuxostat est un dérivé 2-arylthiazole qui exerce son effet thérapeutique de diminution de l'uricémie en inhibant sélectivement la XO. Le fébuxostat est un inhibiteur non purinique puissant et sélectif de la XO (NP-SIXO). In vitro, sa constante d'inhibition Ki est inférieure à une nanomole. Le fébuxostat inhibe de façon puissante les formes oxydée et réduite de la XO. Aux concentrations thérapeutiques, le fébuxostat n'inhibe pas les autres enzymes intervenant dans le métabolisme des purines ou des pyrimidines (guanine désaminase, hypoxanthine guanine phosphoribosyltransférase, orotate phosphoribosyltransférase, orotidine monophosphate décarboxylase ou purine nucléoside phosphorylase).
Résultats des études cliniques :
L'efficacité d'Adenuric a été démontrée au cours de trois études pivots de phase III (les deux études pivots APEX et FACT et l'étude additionnelle CONFIRMS décrites ci-dessous) menées chez 4101 patients présentant une hyperuricémie et une goutte. Au cours de ces 2 études, Adenuric a démontré sa supériorité vis-à-vis de l'allopurinol pour diminuer et maintenir l'uricémie. Le critère principal d'efficacité au cours des études APEX et FACT était la proportion des patients présentant une uricémie < 6,0 mg/dl (357 µmol/l) au cours des 3 dernières mesures mensuelles. Au cours de l'étude additionnelle de phase III CONFIRMS, dont les résultats ont été obtenus après l'octroi de l'autorisation de mise sur le marché, le critère principal d'efficacité était la proportion de patients présentant une uricémie < 6,0 mg/dl à la dernière visite. Aucun patient ayant reçu une greffe d'organe n'a été inclus dans ces études (cf Posologie et Mode d'administration).
Étude APEX :
L'étude APEX (Allopurinol and Placebo-Controlled Efficacy Study of Febuxostat) est une étude de phase III multicentrique randomisée, menée en double insu, d'une durée de 28 semaines contrôlée contre placebo et allopurinol. 1072 patients ont été randomisés dans les groupes suivants : placebo (n = 134), Adenuric 80 mg une fois par jour (n = 267), Adenuric 120 mg une fois par jour (n = 269), Adenuric 240 mg une fois par jour (n = 134) ou allopurinol (300 mg une fois par jour [n = 258] chez les patients dont la créatinémie initiale était <= 1,5 mg/dl ou 100 mg une fois par jour [n = 10] chez ceux dont la créatinémie initiale était > 1,5 mg/dl et <= 2,0 mg/dl). La dose de 240 mg de fébuxostat (deux fois la plus forte dose recommandée) a été étudiée pour évaluer la tolérance.
L'étude APEX a démontré la supériorité statistiquement significative d'Adenuric 80 mg une fois par jour et d'Adenuric 120 mg une fois par jour par rapport à l'allopurinol administré aux doses conventionnelles de 300 mg (n = 258)/100 mg (n = 10) sur la diminution de l'uricémie en dessous du seuil de 6 mg/dl (357 µmol/l) ; voir tableau 2 et figure 1.
Étude FACT :
L'étude FACT (Febuxostat Allopurinol Controlled Trial) est une étude de phase III multicentrique randomisée, menée en double insu, d'une durée de 52 semaines, contrôlée contre allopurinol. Sept cent soixante (760) patients ont été randomisés dans les groupes suivants : Adenuric 80 mg une fois par jour (n = 256), Adenuric 120 mg une fois par jour (n = 251) et allopurinol 300 mg une fois par jour (n = 253).
L'étude FACT a montré la supériorité statistiquement significative d'Adenuric 80 mg une fois par jour et d'Adenuric 120 mg une fois par jour par rapport à l'allopurinol administré à la dose conventionnelle de 300 mg sur la réduction et le maintien de l'uricémie au-dessous du seuil de 6 mg/dl (357 µmol/l).
Le tableau 2 résume les résultats sur le critère principal d'efficacité.
Tableau 2 : Proportion des patients présentant une uricémie < 6,0 mg/dl (357 µmol/l) au cours des trois dernières visites mensuelles :
Étude
Adenuric 80 mg 1 fois/jour
Adenuric 120 mg 1 fois/jour
Allopurinol 300/100 mg 1 fois/jour*
APEX (28 semaines)
48 %(1) (n = 262)
65 %(1)(2) (n = 269)
22 % (n = 268)
APEX (52 semaines)
53 %(1) (n = 255)
62 %(1) (n = 250)
21 % (n = 251)
Résultats regroupés
51 %(1) (n = 517)
63 %(1)(2) (n = 519)
22 % (n = 519)
*
Les résultats observés chez les sujets recevant 100 mg une fois par jour (n = 10, créatininémie > 1,5 et <= 2,0 mg/dl) ou 300 mg une fois par jour (n = 509) ont été regroupés pour les analyses.
(1)
p < 0,001 vs allopurinol.
(2)
p < 0,001 vs 80 mg.
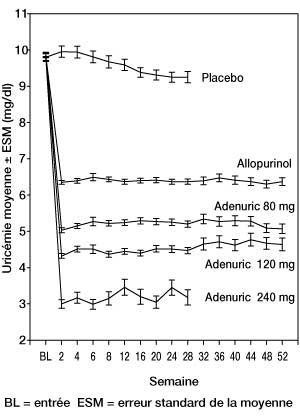
La diminution de l'uricémie sous l'effet d'Adenuric a été rapide et persistante. Une réduction de l'uricémie sous le seuil de 6,0 mg/dl (357 µmol/l) a été notée dès la visite en semaine 2 et s'est maintenue pendant toute la durée du traitement. La figure 1 présente l'évolution de l'uricémie moyenne au cours du temps dans chaque groupe de traitement au cours des deux études pivots de phase III.
Figure 1 : Uricémie moyenne des études pivots de phase III (résultats combinés)
Note : 509 patients ont reçu l'allopurinol à raison de 300 mg 1 fois/jour ; 10 patients dont la créatininémie était > 1,5 et < 2,0 mg/dl ont reçu 100 mg 1 fois/jour (10 patients sur 268 dans l'étude APEX). La dose de 240 mg a été utilisée pour évaluer la tolérance du fébuxostat à une dose deux fois supérieure à la dose maximale recommandée.
Étude CONFIRMS :
L'étude CONFIRMS est une étude de phase III, randomisée, contrôlée, d'une durée de 26 semaines dont l'objectif était d'évaluer la tolérance et l'efficacité du fébuxostat 40 et 80 mg comparativement à l'allopurinol 300 ou 200 mg chez des patients atteints de goutte et présentant une hyperuricémie. 2269 patients ont été randomisés : groupe Adenuric 40 mg une fois par jour (n = 757), groupe Adenuric 80 mg une fois par jour (n = 756), groupe allopurinol 300/200 mg une fois par jour (n = 756). Au moins 65 % des patients avaient une insuffisance rénale légère (clairance de la créatinine comprise entre 30 et 89 ml/min). Une prophylaxie des crises de goutte était obligatoire pendant les 26 semaines de traitement.
La proportion de patients avec une uricémie < 6 mg/dl (357 µmol) à la dernière visite était de 45 % dans le groupe fébuxostat 40 mg, 67 % dans le groupe fébuxostat 80 mg et 42 % dans le groupe allopurinol 300/200 mg.
Critère principal dans le sous-groupe des patients insuffisants rénaux :
L'étude APEX a évalué l'efficacité chez 40 patients insuffisants rénaux (définie par une créatininémie initiale > 1,5 mg/dl et <= 2,0 mg/dl). Chez les insuffisants rénaux randomisés dans le groupe allopurinol, la dose a été limitée à 100 mg une fois par jour. Le critère principal d'efficacité a été atteint sous Adenuric chez 44 % (80 mg une fois par jour), 45 % (120 mg une fois par jour) et 60 % (240 mg une fois par jour) des patients contre 0 % des patients inclus dans le groupe allopurinol 100 mg une fois par jour et dans le groupe placebo.
La diminution de l'uricémie en pourcentage n'a pas différé de façon cliniquement significative en fonction de l'état de la fonction rénale (58 % dans le groupe fonction rénale normale et 55 % dans le groupe dysfonction rénale sévère).
Une analyse, définie de façon prospective dans l'étude CONFIRMS, effectuée chez les patients atteints de goutte présentant une insuffisance rénale légère à modérée (65 % des patients étudiés) a montré que le fébuxostat était significativement plus efficace que l'allopurinol 300/200 mg pour abaisser l'uricémie en deçà de 6 mg/dl.
Critère principal dans le sous-groupe des patients présentant une uricémie >= 10 mg/dl :
L'uricémie initiale était >= 10 mg/dl chez environ 40 % des patients inclus dans les études APEX et FACT (considérées simultanément). Dans ce sous-groupe, le critère principal d'efficacité (uricémie < 6,0 mg/dl aux trois dernières visites) a été atteint sous Adenuric chez 41 % (80 mg une fois par jour), 48 % (120 mg une fois par jour) et 66 % (240 mg une fois par jour) des patients contre 9 % des patients inclus dans le groupe allopurinol 300 mg/100 mg une fois par jour et 0 % dans le groupe placebo.
Au cours de l'étude CONFIRMS (Adenuric 80 mg) : La proportion de patients ayant atteint le critère principal d'efficacité (uricémie < 6 mg/dl à la dernière visite) parmi ceux ayant une uricémie initiale >= 10 mg/dl était de 27 % (66/249) chez les patients traités par fébuxostat 40 mg une fois par jour, 49 % (125/254) chez les patients traités par fébuxostat 80 mg une fois par jour et 31 % (72/230) chez les patients traités par allopurinol 300/200 mg.
Critères cliniques : proportion de patients ayant nécessité un traitement de la crise de goutte :
Étude APEX : Au cours de la période de prophylaxie de 8 semaines, une proportion plus importante de sujets du groupe fébuxostat 120 mg (36 %) a nécessité un traitement de la crise de goutte comparativement aux groupes fébuxostat 80 mg (22 %), allopurinol 300 mg (23 %) et placebo (20 %). Les crises ont augmenté après la période de prophylaxie puis ont diminué graduellement au cours du temps. Entre 46 % et 55 % des sujets ont reçu un traitement de la crise de goutte de la semaine 8 à la semaine 28. Les crises de goutte survenues durant les 4 dernières semaines de l'étude (semaine 24 - semaine 28) ont été observées chez 15 % des sujets du groupe fébuxostat 80/120 mg, 14 % des sujets du groupe allopurinol 300 mg et 20 % des sujets du groupe placebo.
Étude FACT : Au cours de la période de prophylaxie de 8 semaines, une proportion plus importante de sujets du groupe fébuxostat 120 mg (36 %) a nécessité un traitement de la crise de goutte comparativement aux groupes fébuxostat 80 mg (22 %) et allopurinol 300 mg (21 %). Après la période de prophylaxie de 8 semaines, l'incidence des crises a augmenté puis a graduellement diminué au cours du temps (64 % et 70 % des sujets ont reçu un traitement de la crise de goutte de la semaine 8 à la semaine 52). Les crises de goutte survenues durant les 4 dernières semaines de l'étude (semaine 49 - semaine 52) ont été observées chez 6 à 8 % des sujets du groupe fébuxostat 80/120 mg et 11 % des sujets du groupe allopurinol 300 mg.
La proportion des sujets ayant nécessité un traitement de la crise de goutte (études APEX et FACT) a été numériquement plus faible dans les groupes où l'uricémie moyenne après l'entrée dans l'étude avait été < 6,0 mg/dl, < 5,0 mg/dl ou < 4,0 mg/dl que dans le groupe où elle avait été >= 6,0 mg/dl au cours des 32 dernières semaines de traitement (intervalles semaine 20-semaine 24 à semaines 49-52).
Au cours de l'étude CONFIRMS, les proportions de patients ayant nécessité un traitement de la crise de goutte (du 1er jour au 6e mois) étaient de 31 % et 25 % respectivement dans le groupe fébuxostat 80 mg et le groupe allopurinol. Aucune différence n'a été observée entre la proportion de patients ayant nécessité un traitement de la crise de goutte entre le groupe fébuxostat 80 mg et le groupe fébuxostat 40 mg.
Études d'extension en ouvert à long terme :
Étude EXCEL (C02-021) : l'étude EXCEL était une étude d'extension de phase III, d'une durée de 3 ans, effectuée en ouvert, multicentrique, randomisée, contrôlée contre allopurinol, évaluant la tolérance chez les patients qui avaient terminé les études pivots de phase III (APEX ou FACT). Au total 1086 patients ont été inclus : groupe Adenuric 80 mg une fois par jour (n = 649), groupe Adenuric 120 mg une fois par jour (n = 292) et groupe allopurinol 300/100 mg une fois par jour (n = 145). Environ 69 % des patients n'ont pas nécessité de modification de leur traitement pour parvenir à un traitement final stable. Les patients ayant trois mesures d'uricémie consécutives > 6 mg/dl ont été sortis de l'étude. Les niveaux d'uricémie se sont maintenus au cours du temps (91 % et 93 % respectivement des patients traités par fébuxostat 80 mg et 120 mg avaient une uricémie < 6 mg/dl à 36 mois).
Les données recueillies pendant 3 ans ont montré une diminution de l'incidence des crises de goutte, un traitement pour une crise de goutte s'étant avéré nécessaire chez moins de 4 % des patients (plus de 96 % des patients n'ont pas été traités pour une crise de goutte) entre les 16e et 24e mois et entre les 30e et 36e mois.
Respectivement 46 et 38 % des patients ayant un traitement final stable par fébuxostat 80 ou 120 mg une fois par jour ont eu une résolution complète du premier tophus palpable entre la visite initiale et la dernière visite.
L'étude TMX-01-005 (FOCUS) était une étude d'extension de phase II d'une durée de 5 ans, en ouvert, multicentrique, évaluant la tolérance chez les patients qui avaient terminé les 4 semaines de traitement par fébuxostat en double aveugle de l'étude de détermination de doses TMX-00-004. 116 patients ont été inclus et ont été traités par fébuxostat 80 mg une fois par jour. 62 % des patients n'ont pas nécessité d'ajustement de la posologie pour maintenir une uricémie < 6 mg/dl et 38 % des patients ont nécessité une adaptation de la posologie avant d'atteindre un traitement final stable.
La proportion de patients avec une uricémie < 6 mg/dl (357 µmol/l) à la dernière visite était supérieure a 80 % (de 81 a 100 %) pour chacune des doses de fébuxostat.
Au cours des études cliniques de phase III, de légères anomalies du bilan hépatique ont été enregistrées chez des patients (5,0 %) traités par le fébuxostat. Ce pourcentage a été similaire à celui rapporté avec l'allopurinol (4,2 %) : cf Mises en garde et Précautions d'emploi. Au cours des études d'extension ouvertes à long terme, une augmentation du taux de TSH (> 5,5 µUI/ml) a été constatée chez des patients traités au long cours par le fébuxostat (5,5 %) et par l'allopurinol (5,8 %) : cf Mises en garde et Précautions d'emploi.
PHARMACOCINÉTIQUE
Chez des sujets en bonne santé, la concentration plasmatique maximale (Cmax) et l'aire sous la courbe des concentrations plasmatiques en fonction du temps (ASC) du fébuxostat ont augmenté de façon dose-dépendante à la suite de l'administration de doses uniques et répétées de 10 à 120 mg. L'ASC a augmenté de façon plus que proportionnelle pour des doses de fébuxostat allant de 120 à 300 mg. Aucune accumulation notable n'a été observée lors de l'administration de 10 à 240 mg toutes les 24 heures. La demi-vie terminale d'élimination apparente moyenne du fébuxostat (t½) est d'environ 5 à 8 heures.
Des analyses pharmacocinétiques/pharmacodynamiques de population ont été menées chez 211 patients présentant une hyperuricémie et une goutte, qui ont été traités par Adenuric 40 à 240 mg une fois par jour. En règle générale, les paramètres pharmacocinétiques du fébuxostat estimés par ces analyses ont été similaires à ceux déterminés chez les sujets sains, indiquant que ces derniers sont représentatifs pour l'évaluation pharmacocinétique/pharmacodynamique chez les patients atteints de goutte.
Absorption :
L'absorption du fébuxostat est rapide (Tmax = 1,0-1,5 h) et élevée (au moins 84 %). Après des doses orales uniques ou répétées de 80 et 120 mg une fois par jour, la Cmax est respectivement d'environ 2,8-3,2 µg/ml et 5,0-5,3 µg/ml. La biodisponibilité absolue de la formulation comprimé du fébuxostat n'a pas été étudiée.
A la suite de doses orales répétées de 80 mg une fois par jour ou d'une dose unique de 120 mg avec un repas riche en lipide, la Cmax a diminué de respectivement 49 % et 38 %, et l'ASC de 18 % et 16 %. Aucune modification cliniquement significative du pourcentage de diminution de l'uricémie n'a été cependant observée quand ce paramètre a été mesuré (doses répétées de 80 mg). Adenuric peut donc être pris conjointement ou non avec une prise alimentaire.
Distribution :
Le volume apparent de distribution à l'état d'équilibre (Vss/F) du fébuxostat est de 29 à 75 l après des doses orales de 10 à 300 mg. La liaison du fébuxostat aux protéines plasmatiques est d'environ 99,2 % (principalement à l'albumine) et est constante avec les concentrations obtenues avec les doses de 80 et 120 mg. La liaison des métabolites actifs aux protéines plasmatiques est d'environ 82 % à 91 %.
Métabolisme :
Le fébuxostat est fortement métabolisé par conjugaison via le système enzymatique diphosphate glucuronosyltransférase (UDPGT) et par oxydation via le cytochrome P450 (CYP). Quatre métabolites hydroxylés pharmacologiquement actifs ont été identifiés, dont trois ont été décelés dans le plasma chez l'homme. Des études in vitro sur microsomes hépatiques humains ont montré que ces métabolites oxydatifs étaient principalement formés par CYP1A1, CYP1A2, CYP2C8 ou CYP2C9 et que le glycuronide du fébuxostat était principalement formé par UGT 1A1, 1A8 et 1A9.
Élimination :
Le fébuxostat est éliminé par voies hépatique et rénale. Après administration par voie orale d'une dose de 80 mg de fébuxostat marqué au 14C, environ 49 % de la dose a été retrouvé dans l'urine sous forme de fébuxostat inchangé (3 %), d'acyl glycuronide de la substance active (30 %), de ses métabolites oxydatifs connus et de leurs dérivés conjugués (13 %) et d'autres métabolites inconnus (3 %). En dehors de l'excrétion urinaire, près de 45 % de la dose a été retrouvé dans les fèces sous forme de fébuxostat inchangé (12 %), d'acyl glycuronide de la substance active (1 %), de ses métabolites oxydatifs connus et de leurs dérivés conjugués (25 %), et d'autres métabolites inconnus (7%).
Populations particulières :
Insuffisance rénale : après administration de doses répétées de 80 mg d'Adenuric, la Cmax du fébuxostat n'est pas différente entre les patients présentant une insuffisance rénale légère, modérée ou sévère par rapport à des sujets à fonction rénale normale. L'ASC moyenne totale du fébuxostat a été environ 1,8 fois plus élevée chez les patients présentant une dysfonction rénale sévère que chez les sujets à fonction rénale normale (13,2 µg × h/ml contre 7,5 µg × h/ml). La Cmax et l'ASC des métabolites actifs ont été respectivement deux et quatre fois plus élevées. Aucune adaptation de la posologie n'est cependant nécessaire chez les patients présentant une insuffisance rénale légère à modérée.
Insuffisance hépatique : après administration de doses répétées de 80 mg d'Adenuric, la Cmax et l'ASC du fébuxostat et de ses métabolites ne sont pas significativement différentes entre les patients présentant une insuffisance hépatique légère (classe A de Child-Pugh) ou modérée (classe B de Child-Pugh) par rapport à des sujets à fonction hépatique normale. Aucune étude n'a été menée chez des patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh).
Age : après administration répétée d'Adenuric par voie orale, aucune différence significative de l'ASC du fébuxostat n'a été observée entre des sujets âgés et des sujets sains plus jeunes.
Sexe : après administration répétée d'Adenuric par voie orale, la Cmax et l'ASC du fébuxostat ont été plus élevées de respectivement 24 % et 12 % chez les femmes que chez les hommes. La Cmax et l'ASC corrigées en fonction du poids ont été cependant similaires entre les sujets des deux sexes. Aucune adaptation de la dose en fonction du sexe n'est nécessaire.
SÉCURITE PRÉCLINIQUE
Les effets observés lors des études précliniques sont généralement survenus à des expositions supérieures à l'exposition maximale chez l'homme.
Cancérogenèse, mutagenèse, altération de la fertilité :
Chez le rat mâle, une augmentation statistiquement significative des tumeurs de la vessie (papillomes et carcinomes à cellules transitionnelles) n'a été observée qu'en association à des calculs de xanthine dans le groupe recevant une dose élevée (environ 11 fois l'exposition humaine). Aucune augmentation significative d'un autre type de tumeur n'a été observée chez la souris et le rat mâle ou femelle. Ces observations sont considérées comme une conséquence d'une composition de l'urine et d'un métabolisme des purines spécifiques à l'espèce et comme dépourvues de signification en clinique.
Une batterie standard de tests de génotoxicité n'a révélé aucun effet génotoxique biologiquement pertinent du fébuxostat.
Le fébuxostat à des doses orales allant jusqu'à 48 mg/jour n'a montré aucun effet sur la fertilité et la capacité de reproduction chez le rat mâle ou femelle.
Aucun signe d'altération de la fertilité, d'effet tératogène ou d'effet délétère sur le foetus lié au fébuxostat n'a été observé. Une toxicité maternelle a été observée aux doses élevées, accompagnée d'une réduction de l'indice de sevrage et du développement des petits chez le rat à une exposition d'environ 4,3 fois celle observée chez l'homme. Des études de tératogenèse menées chez la rate gestante à une exposition équivalente à environ 4,3 fois l'exposition humaine et chez la lapine gestante à une exposition d'environ 13 fois celle-ci n'ont révélé aucun effet tératogène.