ZYPADHERA®
olanzapine
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p flacon | |
| Olanzapine (DCI) pamoate monohydraté exprimé en olanzapine | 210 mg |
| ou | 300 mg |
| ou | 405 mg |
Poudre : pas d'excipients.
Solvant : croscarmellose sodique, mannitol, polysorbate 80, eau pour injection, acide chlorhydrique et hydroxyde de sodium (pour ajustement du pH).
Après reconstitution, chaque ml de suspension contient 150 mg d'olanzapine.
INDICATIONS |
POSOLOGIE ET MODE D'ADMINISTRATION |
Zypadhera doit être administré uniquement par injection intramusculaire profonde dans le muscle fessier (glutéal) par un professionnel de santé entraîné aux techniques d'injection appropriées et dans un lieu où une surveillance post-injection et un accès à des soins médicaux appropriés en cas de surdosage peuvent être assurés.
Après chaque injection, les patients doivent être surveillés dans un établissement de soin par du personnel qualifié approprié pendant au moins 3 heures afin de détecter les signes et symptômes d'un surdosage par olanzapine. Il faut s'assurer que le patient est bien éveillé, orienté et ne présente pas de signe ou symptôme de surdosage. Si un surdosage est suspecté, une prise en charge et une surveillance médicale étroites doivent être poursuivies jusqu'à ce que l'examen clinique indique que les signes et symptômes du surdosage ont disparu (cf Mises en garde et Précautions d'emploi).
Les patients doivent initialement être traités par olanzapine orale avant d'administrer Zypadhera pour établir la tolérance et la réponse au traitement.
Pour les instructions d'utilisation, cf Modalités de manipulation et d'élimination.
Ne pas confondre Zypadhera 210, 300 ou 405 mg poudre et solvant pour suspension injectable à libération prolongée avec olanzapine 10 mg poudre pour solution injectable.
Se référer au schéma posologique du tableau 1 pour connaître la première dose de Zypadhera pour tous les patients.
| Dose orale cible d'olanzapine | Dose initiale recommandée de Zypadhera | Dose de maintien après 2 mois de traitement par Zypadhera |
| 10 mg/jour | 210 mg/2 semaines ou 405 mg/4 semaines | 150 mg/2 semaines ou 300 mg/4 semaines |
| 15 mg/jour | 300 mg/2 semaines | 210 mg/2 semaines ou 405 mg/4 semaines |
| 20 mg/jour | 300 mg/2 semaines | 300 mg/2 semaines |
- Adaptation posologique :
- Les patients doivent être étroitement surveillés afin de détecter des signes de rechute au cours du premier et du deuxième mois de traitement. Lors d'un traitement antipsychotique, l'amélioration de l'état clinique du patient peut prendre plusieurs jours à quelques semaines. Les patients doivent être étroitement surveillés durant cette période. Pendant le traitement, la dose peut être ajustée en fonction de l'état clinique du patient. Après réévaluation clinique, la dose peut être ajustée dans une fourchette de 150 à 300 mg toutes les 2 semaines ou de 300 à 405 mg toutes les 4 semaines (cf tableau 1).
- Supplémentation :
- Une supplémentation par olanzapine orale n'était pas autorisée lors des essais cliniques en double aveugle. Si une supplémentation par olanzapine orale est cliniquement indiquée, alors la dose totale combinée d'olanzapine des 2 formulations ne doit pas excéder une dose correspondant à une dose maximum de 20 mg/jour d'olanzapine orale.
- Substitution par un autre médicament antipsychotique :
- Il n'y a pas de données collectées de façon systématique pour évaluer spécifiquement la substitution de Zypadhera par un autre traitement antipsychotique. La dissolution lente du sel de pamoate d'olanzapine permet une libération lente continue d'olanzapine qui se termine environ 6 à 8 mois après la dernière injection. La surveillance par un clinicien, en particulier pendant les deux premiers mois après l'arrêt de Zypadhera est nécessaire lors de la substitution par un autre traitement antipsychotique et est considérée comme médicalement appropriée.
- Patients âgés :
- Zypadhera n'a pas été étudié de façon systématique chez les patients âgés (> 65 ans). Zypadhera n'est pas recommandé pour le traitement des patients âgés à moins qu'un traitement bien toléré et efficace par olanzapine orale n'ait été établi. Une dose initiale plus faible (150 mg toutes les 4 semaines) n'est pas indiquée en routine mais doit être envisagée pour les patients de 65 ans et plus lorsque l'état clinique le nécessite. Il n'est pas recommandé d'initier un traitement par Zypadhera chez les sujets de plus de 75 ans (cf Mises en garde et Précautions d'emploi).
- Insuffisants rénaux et/ou hépatiques :
- Zypadhera ne doit pas être utilisé chez ces patients à moins qu'un traitement bien toléré et efficace par olanzapine orale n'ait été établi. Une dose initiale plus faible (150 mg toutes les 4 semaines) doit être envisagée pour ces patients. En cas d'insuffisance hépatique modérée (cirrhose, Child-Pugh de classe A ou B), la dose initiale devra être de 150 mg toutes les 4 semaines et ne sera augmentée qu'avec précaution.
- Sexe :
- La dose initiale et l'intervalle de doses ne nécessitent pas d'adaptation chez la femme par rapport à l'homme.
- Fumeurs :
- La dose initiale et l'intervalle de doses ne nécessitent pas d'adaptation chez les patients non fumeurs par rapport aux fumeurs.
- L'existence de plus d'un facteur pouvant ralentir le métabolisme (sexe féminin, sujet âgé, non fumeur) peut justifier une réduction de la dose. Lorsqu'elle est indiquée, l'augmentation posologique sera faite avec précaution chez ces patients.
- Population pédiatrique :
- Zypadhera n'est pas recommandé chez l'enfant et l'adolescent de moins de 18 ans en raison d'un manque de données de sécurité et d'efficacité.
CONTRE-INDICATIONS |
- Hypersensibilité à la substance active ou à l'un des excipients.
- Patients présentant un risque connu de glaucome à angle fermé.
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
- Utilisation chez les patients agités en phase aiguë ou dans un état psychotique sévère :
- Zypadhera ne doit pas être utilisé pour traiter les patients schizophréniques agités en phase aiguë ou dans un état psychotique sévère nécessitant un contrôle immédiat des symptômes.
- Syndrome post-injection :
- Lors des études cliniques conduites avant la commercialisation, des réactions présentant des signes et symptômes compatibles avec un surdosage en olanzapine ont été rapportées chez des patients à la suite d'une injection de Zypadhera. Ces réactions se sont produites dans moins de 0,1 % des injections et chez environ 2 % des patients. La plupart de ces patients ont développé des symptômes de sédation (allant d'une sédation légère à un coma) et/ou de delirium (incluant confusion, désorientation, agitation, anxiété et autres troubles cognitifs). Les autres symptômes observés incluaient symptômes extrapyramidaux, dysarthrie, ataxie, agressivité, vertiges, faiblesse, hypertension et convulsion. Dans la plupart des cas, les signes et symptômes liés à cette réaction sont apparus dans l'heure qui a suivi l'injection, et dans tous les cas un rétablissement complet a été rapporté dans les 24-72 heures après l'injection. Ces réactions se sont produites rarement (< 1/1000 injections) entre 1 et 3 heures après injection et très rarement (< 1/10 000 injections) plus de 3 heures après injection. Les patients doivent être informés de ce risque potentiel et de la nécessité de rester pendant 3 heures dans un établissement de soins après chaque administration de Zypadhera.
- Avant l'injection, le professionnel de santé doit s'assurer que le patient ne se rendra pas seul vers sa destination. Après chaque injection, les patients doivent rester sous surveillance pendant au moins 3 heures dans un établissement de soins avec du personnel qualifié approprié afin de détecter les signes et les symptômes compatibles avec un surdosage en olanzapine.
- Il faut s'assurer que le patient est bien éveillé, orienté et ne présente pas de signe ou symptôme de surdosage. Si un surdosage est suspecté, une prise en charge et une surveillance médicale étroite doivent être poursuivies jusqu'à ce que l'examen clinique indique la disparition des signes et symptômes du surdosage.
- Le reste de la journée, après l'injection, il faut conseiller au patient d'être attentif à tout signe ou symptôme d'un surdosage secondaire à un effet indésirable post-injection, afin d'avoir la possibilité d'obtenir de l'aide si besoin. Les patients ne doivent pas conduire ou utiliser de machines (cf Conduite et Utilisation de machines).
- Si l'utilisation d'une benzodiazépine parentérale est absolument nécessaire pour la prise en charge des réactions secondaires post-injection, le patient devra être étroitement surveillé afin de détecter une sédation excessive et une dépression cardiorespiratoire (cf Interactions).
- Effets indésirables au site d'injection :
- L'effet indésirable au site d'injection le plus fréquemment rapporté a été la douleur. La majorité de ces réactions était rapportée comme étant « légère » à « modérée ». Si un effet indésirable au site d'injection se produit, des mesures appropriées doivent être prises (cf Effets indésirables).
- Démence accompagnée de troubles psychotiques et/ou troubles du comportement :
- L'olanzapine n'est pas indiquée dans le traitement de la démence accompagnée de troubles psychotiques et/ou troubles du comportement et son utilisation chez ce groupe spécifique de patients est déconseillée du fait d'une augmentation du risque de mortalité et d'accidents vasculaires cérébraux. Au cours d'essais cliniques contrôlés versus placebo (durée de 6 à 12 semaines), réalisés chez des patients âgés (âge moyen 78 ans) souffrant de démence accompagnée de troubles psychotiques et/ou de troubles du comportement, l'incidence des décès dans le groupe olanzapine par voie orale a été deux fois plus importante que celle observée dans le groupe placebo (3,5 % versus 1,5 % respectivement). L'incidence plus élevée de décès n'a pas été corrélée à la dose d'olanzapine (dose moyenne quotidienne de 4,4 mg) ou à la durée de traitement. Dans cette population de patients, un âge supérieur à 65 ans, une dysphagie, une sédation, une malnutrition et une déshydratation, une pathologie pulmonaire (telle qu'une pneumopathie avec ou sans inhalation) ou une utilisation concomitante de benzodiazépines peuvent être des facteurs prédisposant à une augmentation du risque de mortalité. Néanmoins, indépendamment de ces facteurs de risque, l'incidence de mortalité a été supérieure dans le groupe olanzapine par voie orale (comparativement au placebo).
- Des réactions vasculaires cérébrales (telles qu'accidents vasculaires cérébraux, accidents ischémiques transitoires), dont certaines à issue fatale, ont été rapportées dans ces mêmes essais cliniques. Trois fois plus d'événements indésirables vasculaires cérébraux ont été rapportés dans le groupe de patients traités par olanzapine par voie orale comparativement au groupe de patients traités par placebo (1,3 % versus 0,4 % respectivement). Tous les patients traités par olanzapine par voie orale ou par placebo ayant présenté un événement vasculaire cérébral, avaient des facteurs de risque préexistants. Un âge supérieur à 75 ans et une démence de type vasculaire ou mixte ont été identifiés comme des facteurs de risque d'événements indésirables vasculaires cérébraux dans le groupe olanzapine. L'efficacité de l'olanzapine n'a pas été démontrée dans ces essais.
- Maladie de Parkinson :
- L'administration de l'olanzapine à des patients parkinsoniens atteints de psychoses médicamenteuses (agonistes dopaminergiques) est déconseillée. Au cours d'essais cliniques, une aggravation de la symptomatologie parkinsonienne et des hallucinations ont été très fréquemment rapportées et de façon plus fréquente qu'avec le placebo (cf Effets indésirables) ; l'olanzapine orale n'était pas plus efficace que le placebo dans le traitement des symptômes psychotiques. Dans ces essais, les patients devaient être stabilisés en début d'étude avec la posologie minimale efficace du traitement antiparkinsonien (agoniste dopaminergique) et poursuivre le même traitement antiparkinsonien, au même dosage, pendant toute l'étude. La posologie initiale de l'olanzapine orale était de 2,5 mg/jour puis pouvait être ajustée par l'investigateur jusqu'à un maximum de 15 mg/jour.
- Syndrome malin des neuroleptiques (SMN) :
- Le syndrome malin des neuroleptiques (SMN) est un syndrome potentiellement mortel associé aux traitements antipsychotiques. De rares cas rapportés comme syndrome malin des neuroleptiques (SMN) ont également été notifiés sous olanzapine orale. Les signes cliniques du SMN sont l'hyperthermie, la rigidité musculaire, l'altération des facultés mentales, et des signes d'instabilité neurovégétative (instabilité du pouls et de la pression artérielle, tachycardie, hypersudation et troubles du rythme cardiaque). Peuvent s'ajouter des signes tels qu'élévation des CPK, myoglobinurie (rhabdomyolyse) et insuffisance rénale aiguë. Si un patient présente des signes ou des symptômes évoquant un SMN, ou une hyperthermie inexpliquée non accompagnée d'autres signes de SMN, tous les médicaments antipsychotiques, y compris l'olanzapine, doivent être arrêtés.
- Hyperglycémie et diabète :
- De rares cas d'hyperglycémie et/ou de survenue ou exacerbation d'un diabète, associés parfois à une acidocétose ou un coma, avec une issue fatale pour certains cas, ont été rapportés (cf Effets indésirables). Dans certains cas, une prise de poids antérieure, qui pourrait être un facteur prédisposant, a été rapportée. Une surveillance clinique appropriée est souhaitable conformément aux recommandations en vigueur sur les antipsychotiques. Les patients traités par antipsychotiques, incluant Zypadhera, doivent être surveillés afin de détecter les signes et symptômes d'une hyperglycémie (tels que polydipsie, polyurie, polyphagie et faiblesse) et les patients ayant un diabète de type II ou des facteurs de risque de diabète de type II doivent être suivis régulièrement pour surveiller la détérioration du contrôle de la glycémie. Le poids doit être surveillé régulièrement.
- Anomalies lipidiques :
- Des anomalies lipidiques ont été observées chez des patients traités par l'olanzapine au cours d'essais cliniques versus placebo (cf Effets indésirables). Les modifications lipidiques doivent être prises en charge de façon appropriée au plan clinique, notamment chez les patients présentant des troubles lipidiques et chez les patients ayant des facteurs de risque pouvant favoriser le développement de troubles lipidiques. Le bilan lipidique des patients traités par antipsychotiques, incluant Zypadhera, doit être surveillé régulièrement conformément aux recommandations en vigueur sur les antipsychotiques.
- Activité anticholinergique :
- Bien que l'olanzapine ait montré une activité anticholinergique in vitro, l'incidence des effets liés à cette activité a été faible au cours des essais cliniques. Cependant, l'expérience clinique de l'olanzapine étant limitée chez les patients ayant une pathologie associée, la prudence est recommandée lors de sa prescription chez des patients présentant des symptômes d'hypertrophie prostatique, d'iléus paralytique ou de toute autre pathologie en rapport avec le système cholinergique.
- Fonction hépatique :
- Des élévations transitoires et asymptomatiques des aminotransférases (ASAT et ALAT) ont été fréquemment observées, notamment en début de traitement. La prudence s'impose chez les patients présentant une élévation des ALAT et/ou des ASAT, chez les patients présentant des signes et symptômes évocateurs d'une atteinte hépatique, chez les patients atteints d'une insuffisance hépatique avant traitement et chez les patients traités par des médicaments potentiellement hépatotoxiques et un suivi doit être instauré. Dans les cas où une hépatite a été diagnostiquée (comprenant des atteintes hépatiques cytolytiques, cholestatiques ou mixtes), le traitement par olanzapine doit être arrêté.
- Neutropénie :
- La prudence s'impose chez les patients dont le nombre de leucocytes et/ou de neutrophiles est faible quelle qu'en soit la cause, chez les patients recevant des médicaments connus pour induire des neutropénies, chez les patients ayant des antécédents de dépression médullaire ou de myélotoxicité médicamenteuse, chez les patients atteints de dépression médullaire qu'elle soit en relation avec une pathologie intercurrente, une radiothérapie ou une chimiothérapie, et chez les patients atteints d'hyperéosinophilie ou de syndromes myéloprolifératifs. Des neutropénies ont été fréquemment rapportées lors de l'administration concomitante de l'olanzapine et du valproate (cf Effets indésirables).
- Arrêt du traitement :
- Des symptômes aigus tels que sueurs, insomnie, tremblements, anxiété, nausées ou vomissements ont été très rarement rapportés (< 0,01 %) lors de l'arrêt brutal du traitement par olanzapine administrée par voie orale.
- Intervalle QT :
- Au cours des essais cliniques avec olanzapine par voie orale, un allongement du QTc cliniquement significatif (QT corrigé selon la formule de Fridericia [QTcF] >= 500 millisecondes [msec] à n'importe quel moment après l'inclusion chez les patients ayant à l'inclusion un QTcF < 500 msec) a été rapporté de manière peu fréquente (0,1 % à 1 %) chez les patients traités par olanzapine, sans différence significative par rapport au placebo quant aux événements cardiaques associés. Au cours des essais cliniques avec l'olanzapine poudre pour solution injectable ou Zypadhera, l'olanzapine n'a pas été associée avec une augmentation persistante du QT absolu ou du QTc. Cependant, comme avec d'autres antipsychotiques, la prudence est recommandée lors de la coprescription avec des médicaments connus pour allonger l'intervalle QTc notamment chez le sujet âgé ou chez des patients présentant un syndrome de QT long congénital, une insuffisance cardiaque congestive, une hypertrophie cardiaque, une hypokaliémie ou une hypomagnésémie.
- Atteintes thromboemboliques :
- Des atteintes thromboemboliques veineuses ont été très rarement rapportées avec l'olanzapine (< 0,01 %). Il n'a pas été établi de lien de causalité entre la survenue de ces atteintes et le traitement par olanzapine. Cependant, les patients schizophrènes présentant souvent des facteurs de risque thromboembolique veineux, tout facteur de risque potentiel d'atteintes thromboemboliques veineuses (telle l'immobilisation) doit être identifié et des mesures préventives mises en oeuvre.
- Activité générale sur le système nerveux central :
- Compte tenu des principaux effets de l'olanzapine sur le système nerveux central, il faudra être prudent lors de l'association avec d'autres médicaments à action centrale et avec l'alcool. Du fait de son activité antagoniste de la dopamine in vitro, l'olanzapine peut antagoniser les effets des agonistes directs et indirects de la dopamine.
- Convulsions :
- L'olanzapine doit être utilisée avec prudence chez les patients qui ont des antécédents de convulsions ou qui sont placés dans des conditions susceptibles d'abaisser leur seuil convulsif. De rares cas de convulsions ont été rapportés chez les patients traités par olanzapine. Dans la plupart de ces cas, il existait soit des antécédents de convulsions soit des facteurs de risque de convulsions.
- Dyskinésie tardive :
- Dans les études comparatives avec olanzapine de durée inférieure ou égale à un an, la survenue des dyskinésies liées au traitement a été significativement plus faible dans le groupe olanzapine. Cependant, le risque de survenue de dyskinésie tardive augmentant avec la durée de l'exposition, la réduction posologique voire l'arrêt du traitement doivent être envisagés dès l'apparition de signes de dyskinésie tardive. Ces symptômes peuvent provisoirement s'aggraver ou même survenir après l'arrêt du traitement.
- Hypotension orthostatique :
- Une hypotension orthostatique a été rarement observée chez les sujets âgés lors des essais cliniques avec l'olanzapine. Comme avec d'autres antipsychotiques, il est recommandé de mesurer périodiquement la pression artérielle des patients de plus de 65 ans.
- Mort subite d'origine cardiaque :
- Depuis la commercialisation de l'olanzapine, des cas de mort subite d'origine cardiaque ont été rapportés chez les patients traités avec l'olanzapine. Dans une étude observationnelle rétrospective, le risque de mort subite présumée d'origine cardiaque chez les patients traités avec l'olanzapine a été environ le double du risque existant chez les patients ne prenant pas d'antipsychotiques. Dans cette étude, le risque avec l'olanzapine a été comparable au risque avec des antipsychotiques atypiques inclus dans une analyse groupée.
- Population pédiatrique :
- L'olanzapine n'est pas indiquée chez les enfants et les adolescents. Des études réalisées chez des patients âgés de 13 à 17 ans ont montré divers événements indésirables, incluant prise de poids, modification des paramètres métaboliques et élévations des taux sanguins de prolactine. Les effets à long terme associés à ces événements n'ont pas été étudiés et demeurent inconnus (cf Effets indésirables, Pharmacodynamie).
- Utilisation chez les sujets âgés (> 75 ans) :
- Il n'y a pas de données disponibles sur l'utilisation de Zypadhera chez les patients de plus de 75 ans. En raison des modifications biochimiques et physiologiques et de la réduction de la masse musculaire chez ces patients, cette formulation n'est pas recommandée dans ce sous-groupe de patients.
INTERACTIONS |
Population pédiatrique : les études d'interaction ont été conduites seulement chez l'adulte.
Il faut être prudent avec les patients traités par des médicaments susceptibles d'induire une hypotension ou une sédation.
- Interactions potentielles ayant un effet sur l'olanzapine :
- L'olanzapine étant métabolisée par le cytochrome CYP1A2, les produits qui stimulent ou inhibent spécifiquement cette isoenzyme peuvent modifier les paramètres pharmacocinétiques de l'olanzapine.
-
- Induction du CYP1A2 : Le métabolisme de l'olanzapine peut être stimulé par le tabagisme et la carbamazépine, ce qui peut entraîner une diminution des concentrations plasmatiques de l'olanzapine. Seule une augmentation légère à modérée de la clairance de l'olanzapine a été observée. Il est probable que les conséquences cliniques soient limitées, mais une surveillance clinique est recommandée et une augmentation de la posologie de l'olanzapine peut être envisagée, si nécessaire (cf Posologie et Mode d'administration).
- Inhibition du CYP1A2 : Il a été montré que la fluvoxamine, inhibiteur spécifique du CYP1A2, inhibe significativement le métabolisme de l'olanzapine. La fluvoxamine entraîne une augmentation moyenne de la Cmax de l'olanzapine de 54 % chez les femmes non fumeuses, et de 77 % chez les hommes fumeurs. L'augmentation moyenne de l'ASC de l'olanzapine était respectivement de 52 % et de 108 %. Une posologie initiale plus faible de l'olanzapine doit être envisagée chez les patients traités par la fluvoxamine ou tout autre inhibiteur du CYP1A2 comme par exemple la ciprofloxacine. Une diminution de la posologie de l'olanzapine doit être envisagée si un traitement par un inhibiteur du CYP1A2 est instauré.
- Induction du CYP1A2 : Le métabolisme de l'olanzapine peut être stimulé par le tabagisme et la carbamazépine, ce qui peut entraîner une diminution des concentrations plasmatiques de l'olanzapine. Seule une augmentation légère à modérée de la clairance de l'olanzapine a été observée. Il est probable que les conséquences cliniques soient limitées, mais une surveillance clinique est recommandée et une augmentation de la posologie de l'olanzapine peut être envisagée, si nécessaire (cf Posologie et Mode d'administration).
- Avec la fluoxétine (inhibiteur du CYP2D6), des doses uniques d'antiacides (aluminium, magnésium) ou la cimétidine, il n'a pas été retrouvé d'effet significatif sur les paramètres pharmacocinétiques de l'olanzapine.
- Effets potentiels de l'olanzapine sur les autres médicaments :
-
- L'olanzapine peut antagoniser les effets directs et indirects des agonistes dopaminergiques.
- L'olanzapine n'inhibe pas les principales isoenzymes du CYP450 in vitro (c'est-à-dire 1A2, 2D6, 2C9, 2C19, 3A4). Par conséquent, aucune interaction particulière n'est attendue comme cela a pu être vérifié lors d'études in vivo au cours desquelles aucune inhibition du métabolisme des produits actifs suivants n'a été mise en évidence : antidépresseurs tricycliques (représentant principalement la voie du CYP2D6), la warfarine (CYP2C9), la théophylline (CYP1A2), ou le diazépam (CYP3A4 et 2C19).
- Aucune interaction n'a été mise en évidence lors de la prise concomitante de l'olanzapine et du lithium ou du bipéridène.
- Le suivi des taux plasmatiques du valproate n'a pas montré la nécessité d'adapter la posologie du valproate après l'instauration d'un traitement par l'olanzapine.
- L'olanzapine peut antagoniser les effets directs et indirects des agonistes dopaminergiques.
- Activité générale sur le système nerveux central :
-
- La prudence est recommandée chez les patients qui consomment de l'alcool ou qui sont traités par des médicaments dépresseurs du système nerveux central.
- L'utilisation concomitante de l'olanzapine et de médicaments antiparkinsoniens chez les patients atteints de la maladie de Parkinson et de démence est déconseillée (cf Mises en garde et Précautions d'emploi).
- La prudence est recommandée chez les patients qui consomment de l'alcool ou qui sont traités par des médicaments dépresseurs du système nerveux central.
- Intervalle QTc :
- La prudence s'impose si l'olanzapine est administrée de manière concomitante avec des médicaments connus pour allonger l'intervalle QTc (cf Mises en garde et Précautions d'emploi).
GROSSESSE et ALLAITEMENT |
Aucune étude contrôlée spécifique n'a été réalisée chez la femme enceinte. Les patientes doivent être averties de la nécessité d'informer leur médecin de toute grossesse ou désir de grossesse au cours du traitement par l'olanzapine. Cependant, l'expérience chez la femme étant limitée, l'olanzapine ne doit être administrée pendant la grossesse que si les bénéfices potentiels justifient les risques foetaux potentiels.
De très rares cas de tremblements, hypertonie, léthargie et somnolence ont été spontanément rapportés chez des nouveau-nés de mères traitées par l'olanzapine durant le 3e trimestre de grossesse.
Allaitement :
Dans une étude sur l'olanzapine orale chez des femmes volontaires qui allaitaient, l'olanzapine a été retrouvée dans le lait maternel. L'exposition moyenne des nouveau-nés à l'état d'équilibre (en mg/kg) a été estimée à environ 1,8 % de la dose d'olanzapine reçue par la mère (en mg/kg). L'allaitement maternel est donc déconseillé aux patientes en cours de traitement par olanzapine.
CONDUITE et UTILISATION DE MACHINES |
Il faut indiquer aux patients qu'ils ne doivent pas conduire ou utiliser de machines pour le restant de la journée après chaque injection, en raison de la possibilité de survenue d'un syndrome post-injection entraînant des symptômes compatibles avec un surdosage en olanzapine (cf Mises en garde et Précautions d'emploi).
EFFETS INDÉSIRABLES |
Les autres effets indésirables observés chez les patients traités par Zypadhera étaient similaires à ceux observés avec l'olanzapine orale. Au cours des essais cliniques avec Zypadhera, le seul effet indésirable rapporté plus souvent dans le groupe Zypadhera par rapport au groupe placebo avec une différence statistiquement significative était la sédation (Zypadhera 8,2 %, placebo 2,0 %). Une sédation a été rapportée chez 4,7 % de l'ensemble des patients traités par Zypadhera.
Au cours des essais cliniques avec Zypadhera, l'incidence des effets indésirables au site d'injection était d'environ 8 %. L'effet indésirable au site d'injection le plus fréquemment rapporté était la douleur (5 %) ; d'autres effets indésirables rapportés au site d'injection étaient (par ordre de fréquence décroissante) : nodule, érythème, réaction non spécifique au site d'injection, irritation, oedème, contusion, hémorragie et anesthésie. Ces événements se sont produits chez environ 0,1 % à 1,1 % des patients.
Les effets indésirables listés ci-dessous ont été observés après administration orale d'olanzapine, mais peuvent également survenir lors de l'administration de Zypadhera.
- Adultes :
- Les effets indésirables le plus fréquemment rapportés (>= 1 % des patients) au cours des essais cliniques ont été : somnolence, prise de poids, éosinophilie, augmentation des taux de prolactine, de cholestérol, de la glycémie et de la triglycéridémie (cf Mises en garde et Précautions d'emploi), glycosurie, augmentation de l'appétit, sensation vertigineuse, akathisie, parkinsonisme (cf Mises en garde et Précautions d'emploi), dyskinésie, hypotension orthostatique, effets anticholinergiques, élévations transitoires asymptomatiques des aminotransférases (cf Mises en garde et Précautions d'emploi), rash, asthénie, fatigue et oedème.
- La liste des effets indésirables présentés ci-dessous a été établie à partir du recueil des événements indésirables et des examens de laboratoire issus de la notification spontanée et des essais cliniques.
- Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
- Les catégories de fréquence sont définies ainsi : très fréquent (>= 10 %), fréquent (>= 1 %, < 10 %), peu fréquent (>= 0,1 %, < 1 %), rare (>= 0,01 %, < 0,1 %), très rare (< 0,01 %), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
- Affections hématologiques et du système lymphatique :
- Fréquent : Éosinophilie.
- Peu fréquent : Leucopénie, neutropénie.
- Fréquence indéterminée : Thrombocytopénie.
- Fréquent : Éosinophilie.
- Affections du système immunitaire :
- Fréquence indéterminée : Réaction allergique.
- Fréquence indéterminée : Réaction allergique.
- Troubles du métabolisme et de la nutrition :
- Très fréquent : Prise de poids(1).
- Fréquent : Augmentation de la cholestérolémie (2)(3), augmentation de la glycémie(4), augmentation de la triglycéridémie(2)(5), glycosurie, augmentation de l'appétit.
- Fréquence indéterminée : Survenue ou exacerbation d'un diabète, associée parfois à une acidocétose ou un coma, avec une issue fatale pour certains cas (cf Mises en garde et Précautions d'emploi), hypothermie.
- Très fréquent : Prise de poids(1).
- Affections du système nerveux :
- Très fréquent : Somnolence.
- Fréquent : Vertiges, akathisie(6), parkinsonisme(6), dyskinésie(6).
- Fréquence indéterminée : Convulsions avec, dans la plupart des cas, des antécédents de convulsions ou bien des facteurs de risque de convulsions rapportés, syndrome malin des neuroleptiques (cf Mises en garde et Précautions d'emploi), dystonie (incluant des crises oculogyres), dyskinésie tardive, symptômes à l'arrêt du traitement(7).
- Très fréquent : Somnolence.
- Affections cardiaques :
- Peu fréquent : Bradycardie, allongement du QTc (cf Mises en garde et Précautions d'emploi).
- Fréquence indéterminée : Tachycardie/fibrillation ventriculaire, mort subite (cf Mises en garde et Précautions d'emploi).
- Peu fréquent : Bradycardie, allongement du QTc (cf Mises en garde et Précautions d'emploi).
- Affections vasculaires :
- Fréquent : Hypotension orthostatique.
- Fréquence indéterminée : Atteinte thromboembolique (comprenant embolie pulmonaire et thrombose veineuse profonde).
- Fréquent : Hypotension orthostatique.
- Affections gastro-intestinales :
- Fréquent : Effets anticholinergiques légers et transitoires tels que constipation et bouche sèche.
- Fréquence indéterminée : Pancréatite.
- Fréquent : Effets anticholinergiques légers et transitoires tels que constipation et bouche sèche.
- Affections hépatobiliaires :
- Fréquent : Élévations transitoires et asymptomatiques des aminotransférases (ASAT, ALAT), particulièrement en début de traitement (cf Mises en garde et Précautions d'emploi).
- Fréquence indéterminée : Hépatite (comprenant des atteintes hépatiques cytolytiques, cholestatiques ou mixtes).
- Fréquent : Élévations transitoires et asymptomatiques des aminotransférases (ASAT, ALAT), particulièrement en début de traitement (cf Mises en garde et Précautions d'emploi).
- Affections de la peau et du tissu sous-cutané :
- Fréquent : Rash.
- Peu fréquent : Réaction de photosensibilité, alopécie.
- Fréquent : Rash.
- Affections musculosquelettiques et systémiques :
- Fréquence indéterminée : Rhabdomyolyse.
- Fréquence indéterminée : Rhabdomyolyse.
- Affections du rein et des voies urinaires :
- Peu fréquent : Incontinence urinaire.
- Fréquence indéterminée : Dysurie.
- Peu fréquent : Incontinence urinaire.
- Affections des organes de reproduction et du sein :
- Fréquence indéterminée : Priapisme.
- Fréquence indéterminée : Priapisme.
- Troubles généraux et anomalies au site d'administration :
- Fréquent : Asthénie, fatigue, oedème.
- Fréquent : Asthénie, fatigue, oedème.
- Investigations :
- Très fréquent : Augmentation de la prolactinémie(8).
- Peu fréquent : Élévation de la créatine phosphokinase, augmentation de la bilirubine totale.
- Fréquence indéterminée : Augmentation des phosphatases alcalines.
- Très fréquent : Augmentation de la prolactinémie(8).
-
(1)
Une prise de poids cliniquement significative a été observée dans toutes les catégories d'indice de masse corporelle (IMC) de départ. Après un traitement de courte durée (durée médiane de 47 jours), une augmentation de poids supérieure ou égale à 7 % par rapport au poids initial a été très fréquente (22 %), une augmentation de poids supérieure ou égale à 15 % par rapport au poids initial a été fréquente (4,2 %) et une augmentation de poids supérieure ou égale à 25 % par rapport au poids initial a été peu fréquente (0,8 %).
Une augmentation de poids supérieure ou égale à 7 %, à 15 % et à 25 % par rapport au poids initial a été très fréquente (64,4 %, 31,7 % et 12,3 % respectivement) lors d'une utilisation prolongée (au moins 48 semaines).
-
(2)
Les augmentations moyennes des taux lipidiques à jeun (cholestérol total, cholestérol LDL et triglycérides) ont été plus élevées chez les patients sans signes de trouble des lipides au début du traitement.
-
(3)
Observée pour des taux à jeun normaux au début du traitement (< 5,17 mmol/l) qui sont devenus élevés (>= 6,2 mmol/l). Une augmentation des taux de cholestérol total à jeun ayant une valeur limite au début du traitement (>= 5,17 mmol - < 6,2 mmol/l) à des valeurs élevées (>= 6,2 mmol/l) a été très fréquente.
-
(4)
Observée pour des taux à jeun normaux au début du traitement (< 5,56 mmol/l) qui sont devenus élevés (>= 7 mmol/l). Une augmentation des taux de glucose à jeun ayant une valeur limite au début du traitement (>= 5,56 mmol - < 7 mmol/l) à des valeurs élevées (>= 7 mmol/l) a été très fréquente.
-
(5)
Observée pour des taux à jeun normaux au début du traitement (< 1,69 mmol/l) qui sont devenus élevés (>= 2,26 mmol/l). Une augmentation des taux de triglycérides à jeun ayant une valeur limite au début du traitement (>= 1,69 mmol - < 2,26 mmol/l) à des valeurs élevées (>= 2,26 mmol/l) a été très fréquente.
-
(6)
Au cours d'essais cliniques, l'incidence des troubles parkinsoniens et des dystonies dans le groupe olanzapine était numériquement supérieure à celle du groupe placebo (pas de différence statistique significative). Les patients traités par l'olanzapine ont présenté une plus faible incidence de troubles parkinsoniens, d'akathisie et de dystonie que les patients traités par l'halopéridol à des posologies comparables. En l'absence d'information précise concernant les antécédents de mouvements anormaux extrapyramidaux de survenue aiguë ou tardive, on ne peut conclure à ce jour que l'olanzapine entraîne moins de dyskinésies tardives et/ou de syndromes extrapyramidaux tardifs.
-
(7)
Des symptômes aigus tels que sueurs, insomnie, tremblements, anxiété, nausées et vomissements ont été rapportés lors de l'arrêt brutal du traitement par olanzapine.
-
(8)
Dans des études cliniques allant jusqu'à 12 semaines, une prolactinémie dépassant la limite supérieure de la normale a été observée chez environ 30 % des patients traités avec l'olanzapine et ayant un taux de prolactine normal au début du traitement. Chez la majorité de ces patients, les augmentations étaient généralement légères et sont restées inférieures à deux fois la limite supérieure de la normale. Généralement, chez les patients traités avec l'olanzapine, les répercussions cliniques potentiellement associées au niveau mammaire et sur les cycles menstruels (par exemple aménorrhée, tension mammaire, galactorrhée chez les femmes et gynécomastie/tension mammaire chez les hommes) ont été peu fréquentes. Des réactions indésirables potentiellement associées à la fonction sexuelle (par exemple dysfonction érectile chez les hommes et diminution de la libido chez les femmes et les hommes) ont été fréquemment observées.
-
- Utilisation prolongée (au moins 48 semaines) :
- La proportion de patients ayant présenté des modifications indésirables cliniquement significatives de la prise de poids, du glucose, du cholestérol total/HDL/LDL ou des triglycérides a augmenté au cours du temps. Chez les patients adultes qui ont suivi 9-12 mois de traitement, le taux d'augmentation de la glycémie sanguine moyenne a diminué après 6 mois environ.
-
- Information complémentaire concernant des populations particulières :
- Au cours d'essais cliniques chez des patients âgés déments, le traitement par olanzapine a été associé à une incidence supérieure de décès et d'événements indésirables vasculaires cérébraux par rapport au placebo (cf Mises en garde et Précautions d'emploi). Une démarche anormale et des chutes ont été des événements indésirables très fréquemment rapportés avec olanzapine. Des pneumopathies, une augmentation de la température corporelle, une léthargie, un érythème, des hallucinations visuelles et des incontinences urinaires ont été fréquemment observés.
- Au cours d'essais cliniques menés chez des patients parkinsoniens souffrant de psychoses médicamenteuses (agonistes dopaminergiques), une aggravation de la symptomatologie parkinsonienne et des hallucinations ont été très fréquemment rapportées, et ce, de façon plus fréquente qu'avec le placebo.
- Au cours d'un essai clinique mené chez des patients présentant un épisode maniaque dans le cadre de troubles bipolaires, lors de la prise concomitante de valproate, la fréquence des neutropénies a été de 4,1 % ; un facteur contributif potentiel pourrait être des taux plasmatiques élevés de valproate. Une augmentation supérieure à 10 % des cas de tremblements, bouche sèche, augmentation de l'appétit et prise de poids a été observée lors de l'association de l'olanzapine au lithium ou au valproate. Des troubles de l'élocution ont également été fréquemment rapportés. Lors de l'association de l'olanzapine au lithium ou au valproate une augmentation supérieure ou égale à 7 % du poids initial est survenue chez 17,4 % des patients pendant la phase aiguë du traitement (jusqu'à 6 semaines). Lors du traitement au long cours par l'olanzapine (jusqu'à 12 mois) dans la prévention des récidives chez les patients présentant un trouble bipolaire, une augmentation de poids supérieure ou égale à 7 % par rapport au poids initial a été rapportée chez 39,9 % des patients.
- Population pédiatrique :
- L'olanzapine n'est pas indiquée chez les enfants et adolescents âgés de moins de 18 ans. Bien qu'aucune étude clinique comparant les adolescents aux adultes n'ait été réalisée, les données issues des études réalisées chez l'adolescent ont été comparées à celles issues des essais chez l'adulte. La liste ci-dessous résume les effets indésirables rapportés avec une fréquence plus importante chez les patients adolescents (âgés de 13 à 17 ans) que chez les patients adultes ou les effets indésirables uniquement observés lors des essais cliniques de courte durée réalisés chez les patients adolescents. Une prise de poids cliniquement significative (>= 7 %) surviendrait plus fréquemment chez les adolescents comparés à des patients adultes avec une exposition comparable. L'amplitude de la prise de poids et la proportion des patients adolescents qui ont eu une augmentation du poids cliniquement significative ont été plus importantes lors d'une exposition prolongée (au moins 24 semaines) que lors d'une exposition de courte durée.
- Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante. Les catégories de fréquence sont définies ainsi : très fréquent (>= 10 %), fréquent (>= 1 %, < 10 %).
- Troubles du métabolisme et de la nutrition :
- Très fréquent : Prise de poids(9), augmentation de la triglycéridémie(10), augmentation de l'appétit.
- Fréquent : Augmentation de la cholestérolémie(11).
- Très fréquent : Prise de poids(9), augmentation de la triglycéridémie(10), augmentation de l'appétit.
- Affections du système nerveux :
- Très fréquent : Sédation (dont hypersomnie, léthargie, somnolence).
- Très fréquent : Sédation (dont hypersomnie, léthargie, somnolence).
- Affections gastro-intestinales :
- Fréquent : Bouche sèche.
- Fréquent : Bouche sèche.
- Affections hépatobiliaires :
- Très fréquent : Élévations des aminotransférases (ASAT, ALAT ; cf Mises en garde et Précautions d'emploi).
- Très fréquent : Élévations des aminotransférases (ASAT, ALAT ; cf Mises en garde et Précautions d'emploi).
- Investigations :
- Très fréquent : Diminution de la bilirubine totale, augmentation de la gamma glutamyl transférase, augmentation de la prolactinémie(12).
- Très fréquent : Diminution de la bilirubine totale, augmentation de la gamma glutamyl transférase, augmentation de la prolactinémie(12).
-
(9)
Après un traitement de courte durée (durée médiane de 22 jours), une augmentation de poids supérieure ou égale à 7 % par rapport au poids initial (kg) a été très fréquente (40,6 %), une augmentation de poids supérieure ou égale à 15 % par rapport au poids initial a été fréquente (7,1 %) et une augmentation de poids supérieure ou égale à 25 % par rapport au poids initial a été fréquente (2,5 %).
Lors d'une exposition prolongée (au moins 24 semaines), 89,4 % des patients ont eu une augmentation de poids supérieure ou égale à 7 %, 55,3 % ont eu une augmentation de poids supérieure ou égale à 15 % et 29,1 % ont eu une augmentation du poids supérieure ou égale à 25 % par rapport à leur poids initial.
-
(10)
Observée pour des taux à jeun normaux au début du traitement (< 1,016 mmol/l) qui sont devenus élevés (>= 1,467 mmol/l) et des modifications des taux de triglycérides à jeun ayant une valeur limite au début du traitement (>= 1,016 mmol - < 1,467 mmol/l) devenant élevée (>= 1,467 mmol/l).
-
(11)
Des modifications des taux de cholestérol total à jeun ayant une valeur normale au début du traitement (< 4,39 mmol) devenant élevée (>= 5,17 mmol/l) a été fréquente. Des modifications des taux de cholestérol total à jeun ayant une valeur limite au début du traitement (>= 4,39 mmol - < 5,17 mmol/l) devenant élevée (>= 5,17 mmol/l) ont été très fréquentes.
-
(12)
Augmentation de la prolactinémie rapportée chez 47,4 % des patients adolescents.
SURDOSAGE |
Bien qu'un surdosage soit moins probable avec un traitement par voie parentérale qu'avec un traitement par voie orale, des informations de référence portant sur un surdosage par olanzapine orale sont indiquées ci-dessous :
- Signes et symptômes :
- En cas de surdosage, les symptômes très fréquemment observés (incidence > 10 %) sont : tachycardie, agitation/agressivité, dysarthrie, symptômes extrapyramidaux divers et diminution du niveau de conscience allant de la sédation au coma.
- Les autres effets cliniquement significatifs du surdosage sont : délire, convulsions, coma, éventuel syndrome malin des neuroleptiques, insuffisance respiratoire, fausse-route, hypertension ou hypotension, arythmies cardiaques (moins de 2 % des cas de surdosage) et arrêt cardiorespiratoire. Des évolutions fatales ont été rapportées pour des surdosages aigus à une dose aussi basse que 450 mg mais une évolution favorable a également été rapportée à la suite d'un surdosage par environ 2 g d'olanzapine orale.
- Traitement du surdosage :
- Il n'y a pas d'antidote spécifique de l'olanzapine. Un traitement symptomatique et une surveillance des fonctions vitales doivent être mis en oeuvre selon l'état clinique, y compris un traitement de l'hypotension et du collapsus circulatoire et une assistance respiratoire. Ne pas utiliser l'adrénaline, la dopamine ou un autre bêta-sympathomimétique car la stimulation des récepteurs bêta adrénergiques peut aggraver l'hypotension. Un monitoring cardiovasculaire est nécessaire pour déceler d'éventuelles arythmies. Une surveillance médicale étroite et le monitoring doivent être poursuivis jusqu'à la guérison du patient.
PHARMACODYNAMIE |
Classe pharmacothérapeutique : diazépines, oxazépines et thiazépines (code ATC : N05AH03).
L'olanzapine est un agent antipsychotique, un traitement antimaniaque et thymorégulateur avec un large profil pharmacologique sur un certain nombre de récepteurs.
Dans les études précliniques, l'olanzapine a montré une affinité pour certains récepteurs (Ki < 100 nM) tels que les récepteurs sérotoninergiques 5-HT2A/2C, 5-HT3, 5-HT6 ; dopaminergiques D1, D2, D3, D4, D5 ; muscariniques cholinergiques M1-M5 ; alpha1 adrénergiques et histaminiques H1. Des études de comportement chez l'animal ont montré un antagonisme des systèmes 5-HT, dopaminergiques et cholinergiques, ce qui confirme le profil de liaison aux récepteurs. Il a été démontré dans des études in vitro que l'olanzapine avait une plus grande affinité pour les récepteurs sérotoninergiques 5-HT2 que pour les récepteurs dopaminergiques D2, et une plus grande activité in vivo sur les modèles 5-HT2, par rapport aux modèles D2. Il a été démontré par des études électrophysiologiques que l'olanzapine réduit de façon sélective la transmission au niveau des neurones dopaminergiques du système mésolimbique (A10) alors que l'effet observé sur le système striatal (A9) impliqué dans l'activité motrice est limité. L'olanzapine réduit la réponse d'évitement conditionné, test qui peut indiquer une activité antipsychotique, à des doses inférieures à celles responsables d'induction de catalepsie, effet qui peut indiquer la survenue d'effets indésirables moteurs. Contrairement à d'autres agents antipsychotiques, l'olanzapine augmente la réponse à un test d' « anxiolyse ».
L'efficacité de Zypadhera dans le traitement et au cours du traitement de maintien de la schizophrénie est cohérente avec l'efficacité établie de la formulation orale d'olanzapine.
Dans une étude de tomographie par émission de positron (PET) chez des patients traités par Zypadhera (300 mg/4 semaines), l'occupation moyenne des récepteurs D2 était de 60 % ou plus à l'issue d'une période de 6 mois (niveau cohérent avec les données de l'olanzapine orale).
Un total de 1469 patients schizophrènes ont été inclus dans 2 études pivotales. La première était une étude contrôlée versus placebo, de 8 semaines, conduite chez des patients adultes (n = 404) qui souffraient de symptômes psychotiques aigus. Les patients étaient randomisés pour recevoir des injections de Zypadhera 405 mg/4 semaines, 300 mg/2 semaines, 210 mg/2 semaines ou placebo/2 semaines. Aucune supplémentation par antipsychotique oral n'était autorisée. Le score PANSS total (Positive and Negative Symptom Scores) a montré une amélioration significative entre les valeurs initiales (score PANSS total moyen initial 101) et les valeurs finales (modification moyenne - 22,57, - 26,32, - 22,49 respectivement) pour chaque dose de Zypadhera (405 mg/4 semaines, 300 mg/2 semaines et 210 mg/2 semaines) comparées au placebo (modification moyenne - 8,51). Le changement moyen entre les valeurs initiales et les valeurs finales indiquait qu'au jour 3, les patients des groupes 300 mg/2 semaines et 405 mg/4 semaines ont eu des réductions plus importantes statistiquement significatives du score PANSS total par rapport au placebo (- 8,6, - 8,2 et - 5,2 respectivement). Les 3 groupes de traitement Zypadhera ont montré une amélioration statistiquement significative supérieure au placebo dès la fin de la semaine 1. Ces résultats montrent une efficacité de Zypadhera au cours des 8 semaines de traitement et un effet dès la première semaine après initiation du traitement.
La seconde était une étude au long cours chez des patients cliniquement stables (n = 1065), score PANSS total moyen à l'inclusion 54,33 à 57,75, initialement traités par olanzapine orale pendant 4 à 8 semaines puis dont le traitement a été modifié soit pour de l'olanzapine orale soit pour Zypadhera pendant 24 semaines. Aucune supplémentation par antipsychotique oral n'était autorisée. Les 2 groupes de traitement 150 mg et 300 mg toutes les 2 semaines (doses groupées pour l'analyse) et 405 mg toutes les 4 semaines ont été non inférieurs aux doses combinées de 10, 15 et 20 mg d'olanzapine orale (doses groupées pour l'analyse) sur le taux d'exacerbations des symptômes de schizophrénie (taux d'exacerbation de 10 %, 10 % et 7 % respectivement). L'exacerbation a été mesurée par l'aggravation des items du score PANSS dérivé de l'échelle BPRS Positive ainsi que par les hospitalisations dues à l'aggravation de symptômes psychotiques positifs. Le groupe de traitement combinant 150 mg et 300 mg/2 semaines a été non inférieur au groupe de traitement 405 mg/4 semaines (taux d'exacerbation de 10 % dans chaque groupe) à 24 semaines après la randomisation.
- Population pédiatrique :
- Zypadhera n'a pas été étudiée dans la population pédiatrique. Les données disponibles chez les adolescents (âgés de 13 à 17 ans) sont limitées à des données d'efficacité à court terme avec olanzapine orale dans la schizophrénie (6 semaines) et la manie associée à des troubles bipolaires de type I (3 semaines), impliquant moins de 200 adolescents. L'olanzapine orale a été utilisée à une dose flexible démarrant à 2,5 mg et allant jusqu'à 20 mg par jour. Durant le traitement par l'olanzapine par voie orale, les adolescents ont pris de manière significative plus de poids comparativement aux adultes. L'ampleur des modifications des taux à jeun de cholestérol total, de triglycérides, de cholestérol LDL et de prolactine (cf Mises en garde et Précautions d'emploi, Effets indésirables) était plus importante chez les adolescents que chez les adultes. Il n'y a pas de données sur le traitement de maintien et les données sur la sécurité à long terme sont limitées (cf Mises en garde et Précautions d'emploi, Effets indésirables).
PHARMACOCINÉTIQUE |
L'olanzapine est métabolisée dans le foie par conjugaison et oxydation. Le principal métabolite circulant est le 10-N-glucuronide ; il ne franchit pas la barrière hémato-encéphalique. Les cytochromes P450-CYP1A2 et P450-CYP2D6 entraînent la formation du métabolite N-desmethyl et du métabolite 2-hydroxymethyl. Ces deux métabolites ont montré une activité pharmacologique in vivo significativement plus faible que l'olanzapine dans les études animales. L'activité pharmacologique principale est due à la molécule mère, l'olanzapine.
Après une injection unique IM de Zypadhera, la lente dissolution du sel de pamoate d'olanzapine dans le tissu musculaire débute immédiatement et permet une libération lente continue de l'olanzapine pendant plus de 4 semaines. La libération diminue dans un délai de 8 à 12 semaines. Une supplémentation antipsychotique n'est pas requise à l'initiation du traitement par Zypadhera (cf Posologie et Mode d'administration).
La combinaison du profil de libération et de la posologie (injection IM toutes les 2 ou 4 semaines) permet de maintenir la concentration plasmatique d'olanzapine. Les concentrations plasmatiques restent mesurables plusieurs mois après chaque injection de Zypadhera. La demi-vie de l'olanzapine après injection de Zypadhera est de 30 jours alors qu'après administration orale elle est de 30 heures. L'absorption et l'élimination sont complètes environ six à huit mois après la dernière injection.
L'olanzapine orale est rapidement distribuée. Le taux de fixation de l'olanzapine aux protéines plasmatiques est d'environ 93 % pour une fourchette de concentration allant d'environ 7 à 1000 ng/ml. L'olanzapine se lie essentiellement à l'albumine et à l'alpha1-glycoprotéine acide.
La clairance plasmatique de l'olanzapine orale est plus faible chez les femmes (18,9 l/h) que chez les hommes (27,3 l/h) et chez les non-fumeurs (18,6 l/h) par rapport aux fumeurs (27,7 l/h). Des différences pharmacocinétiques similaires ont été observées entre les femmes et les hommes et les fumeurs et les non-fumeurs au cours des essais cliniques avec Zypadhera. Toutefois, l'impact du sexe ou du tabagisme sur la clairance de l'olanzapine est faible par rapport à la variabilité globale interindividuelle.
Après injections intramusculaires répétées avec 150 à 300 mg de Zypadhera toutes les deux semaines, le 10e au 90e percentile de l'état d'équilibre des concentrations plasmatiques d'olanzapine étaient entre 4,2 et 73,2 ng/ml. Les concentrations plasmatiques de l'olanzapine observées dans l'intervalle de dose de 150 mg/4 semaines et 300 mg/2 semaines ont montré une augmentation de l'exposition systémique à l'olanzapine avec l'augmentation des doses de Zypadhera. Lors des 3 premiers mois de traitement avec Zypadhera, l'accumulation de l'olanzapine a été observée mais il n'y a pas eu d'accumulation supplémentaire lors d'une utilisation au long cours (12 mois) chez les patients qui ont reçu des injections allant jusqu'à 300 mg/2 semaines.
Il n'y a pas eu d'études spécifiques chez le sujet âgé avec Zypadhera. Zypadhera n'est pas recommandé chez les sujets âgés (65 ans et plus) à moins qu'un schéma posologique bien toléré et efficace n'ait été établi avec l'olanzapine orale. Chez le sujet âgé sain (65 ans et plus) par rapport au sujet jeune, la demi-vie d'élimination moyenne a été prolongée (51,8 h versus 33,8 h) et la clairance a été diminuée (17,5 l/h versus 18,2 l/h). La variabilité pharmacocinétique observée chez le sujet âgé est du même ordre que celle du sujet jeune. Chez 44 patients schizophrènes de plus de 65 ans, des doses de 5 à 20 mg/jour n'ont pas été associées avec des effets indésirables différents.
Chez les patients atteints d'insuffisance rénale (clairance de la créatinine < 10 ml/min), par rapport aux sujets sains, ni la demi-vie d'élimination moyenne (37,7 h versus 32,4 h), ni la clairance (21,2 l/h versus 25,0 l/h) ne sont significativement différentes. Toutefois, des études du bilan de masse ont montré qu'environ 57 % d'une dose d'olanzapine marquée par un isotope radioactif ont été excrétés dans les urines, principalement sous forme de métabolites. Bien que Zypadhera n'ait pas été étudié chez les patients atteints d'insuffisance rénale, il est recommandé qu'un schéma posologique bien toléré et efficace ait été établi avec l'olanzapine orale avant d'initier un traitement par Zypadhera (cf Posologie et Mode d'administration).
Chez les sujets fumeurs avec une insuffisance hépatique modérée recevant l'olanzapine par voie orale, la demi-vie d'élimination moyenne est prolongée (39,3 h) et la clairance (18,0 l/h) est réduite de la même façon que chez les sujets sains non fumeurs (respectivement 48,8 h et 14,1 l/h). Bien que Zypadhera n'ait pas été étudié chez les patients atteints d'insuffisance hépatique, il est recommandé qu'un schéma posologique bien toléré et efficace ait été établi avec l'olanzapine orale avant d'initier un traitement par Zypadhera (cf Posologie et Mode d'administration).
Une étude avec l'olanzapine orale comprenant des sujets caucasiens, japonais et chinois, n'a montré aucune différence dans les paramètres pharmacocinétiques entre les trois populations.
SÉCURITE PRÉCLINIQUE |
Des études de sécurité préclinique ont été réalisées avec le pamoate monohydraté d'olanzapine. Les principaux événements retrouvés au cours de ces études de toxicité à doses répétées (rat, chien), dans une étude de carcinogénicité de 2 ans chez le rat, et dans des études de toxicité de la reproduction (rat, lapin) ont été limités à des réactions au site d'injection pour lesquelles aucun NOAEL (No Observed Adverse Effect Level) n'a pu être déterminé. Aucun nouvel effet toxique résultant d'une exposition systémique à l'olanzapine n'a pu être identifié. Cependant, les concentrations systémiques dans ces études étaient généralement plus faibles que celles auxquelles des effets ont été vus dans les études par voie orale ; par conséquent, l'information sur l'olanzapine orale est indiquée ci-dessous pour référence.
- Toxicité aiguë (dose unique) :
- Les signes de toxicité après administration orale chez les rongeurs sont caractéristiques des neuroleptiques puissants : hypoactivité, coma, tremblements, convulsions cloniques, hypersalivation et diminution de la prise de poids. Les doses médianes létales étaient d'environ 210 mg/kg (souris) et 175 mg/kg (rats). Les chiens ont toléré des doses orales uniques allant jusqu'à 100 mg/kg sans décéder. Les signes cliniques observés ont été les suivants : sédation, ataxie, tremblements et accélération de la fréquence cardiaque, respiration difficile, myosis et anorexie. Chez le singe, des doses orales uniques allant jusqu'à 100 mg/kg ont entraîné une prostration et, à des doses supérieures, un état de semi-inconscience.
- Toxicité à doses répétées :
- Dans des études d'une durée allant jusqu'à 3 mois chez la souris, et jusqu'à 1 an chez le rat et le chien, les effets essentiels ont été une dépression du SNC, des effets anticholinergiques et des troubles hématologiques périphériques. Une tolérance est apparue pour la dépression du SNC. Les paramètres de croissance ont été diminués aux fortes doses. Les effets réversibles liés à l'augmentation de la prolactinémie chez la rate comprenaient une diminution du poids des ovaires et de l'utérus, des modifications morphologiques de l'épithélium vaginal et de la glande mammaire.
- Toxicité hématologique :
- Des effets hématologiques ont été observés dans chacune des espèces, y compris des diminutions dose-dépendantes du nombre des leucocytes circulants chez la souris et une diminution non spécifique des leucocytes circulants chez le rat ; cependant, aucun signe de cytotoxicité médullaire n'a été mis en évidence. Une neutropénie réversible, une thrombocytopénie périphérique ou une anémie sont survenues chez quelques chiens traités par 8 ou 10 mg/kg/j (l'exposition totale à l'olanzapine [ASC] étant 12 à 15 fois plus élevée que celle d'un homme ayant reçu une dose de 12 mg). Chez des chiens cytopéniques, aucun effet indésirable sur les cellules souches ou prolifératives de la moelle osseuse n'a été observé.
- Toxicité de reproduction :
- L'olanzapine n'a montré aucun effet tératogène. La sédation a eu un effet sur la capacité d'accouplement des rats mâles. Les cycles oestraux ont été affectés aux doses de 1,1 mg/kg (soit 3 fois la posologie maximale chez l'homme) et les paramètres de reproduction ont été influencés chez les rats ayant reçu des doses de 3 mg/kg (9 fois la posologie maximale chez l'homme). Dans les portées de rats ayant reçu de l'olanzapine, un retard du développement foetal et une diminution transitoire du taux d'activité des petits ont été observés.
- Mutagénicité :
- L'olanzapine n'a montré aucun effet mutagène ni clastogène lors d'une série complète de tests standards, tels que tests de mutation bactérienne et tests in vitro et in vivo sur mammifères.
- Carcinogénicité :
- D'après les résultats des études par voie orale chez la souris et le rat, il a été conclu que l'olanzapine n'est pas carcinogène.
INCOMPATIBILITÉS |
Ce médicament ne doit pas être mélangé avec d'autres médicaments, à l'exception de ceux mentionnés dans la rubrique Modalités de manipulation/Élimination.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 2 ans.
Ne pas réfrigérer ou congeler.
- Après reconstitution dans le flacon :
- 24 heures. Si le produit n'est pas utilisé immédiatement, il doit être vigoureusement mélangé pour être remis en suspension. Une fois que la suspension a été transférée du flacon dans la seringue, elle doit être utilisée immédiatement.
- La stabilité physico-chimique de la suspension dans le flacon a été démontrée pendant 24 h à 20-25 °C. D'un point de vue microbiologique, le produit doit être utilisé immédiatement. S'il n'est pas utilisé immédiatement, les conditions et la durée de conservation avant utilisation sont de la responsabilité de l'utilisateur et ne doivent normalement pas dépasser 24 h à 20-25 °C.
MODALITÉS MANIPULATION/ÉLIMINATION |
Pour injection intramusculaire profonde dans le fessier (glutéal) seulement. Ne pas administrer par voie intraveineuse ou sous-cutanée.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
- Reconstitution :
-
- Étape 1 : Préparation du matériel :
- Il est recommandé d'utiliser des gants car Zypadhera peut irriter la peau.
- Reconstituer Zypadhera poudre pour suspension injectable à libération prolongée uniquement avec le solvant fourni dans le conditionnement en utilisant les techniques d'asepsie standards pour la reconstitution des produits à usage parentéral.
-
- Étape 2 : Détermination du volume de solvant pour la reconstitution :
- Ce tableau indique les quantités nécessaires de solvant pour reconstituer Zypadhera poudre pour suspension injectable à libération prolongée.
-
Dosage du flacon de Zypadhera (mg) Volume de solvant à ajouter (ml) 210 1,3 300 1,8 405 2,3 - Il est important de noter qu'il y a plus de solvant dans le flacon que nécessaire pour la reconstitution.
-
- Étape 3 : Reconstitution de Zypadhera :
-
- Tapoter légèrement le flacon pour « aérer » la poudre.
- Ouvrir la seringue hypodermique Needle-Pro préemballée et l'aiguille avec le dispositif de protection de l'aiguille. Ouvrir la plaquette et enlever le dispositif. S'assurer que l'aiguille est fermement fixée sur le dispositif Needle-Pro en appuyant tout en tournant dans le sens des aiguilles d'une montre, puis retirer le capuchon protecteur de l'aiguille. Le non-respect de ces instructions peut entraîner des blessures avec l'aiguille.
- Prélever dans la seringue le volume de solvant prédéterminé (Étape 2).
- Injecter ce volume de solvant dans le flacon de poudre.
- Enlever l'air pour équilibrer la pression dans le flacon.
- Retirer l'aiguille, en tenant le flacon vers le haut pour éviter toute perte de solvant.
- Fixer le dispositif de sécurité de l'aiguille. Remettre à l'aiguille son capuchon protecteur en utilisant la même main que celle qui tient la seringue. Appuyer doucement la protection contre une surface plane avec la même main. Une fois que la protection est enfoncée, l'aiguille est fermement maintenue dans son capuchon protecteur (figures 1 et 2).
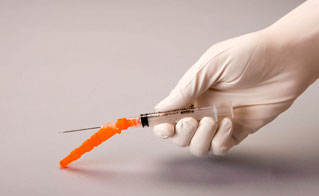
Figure 1
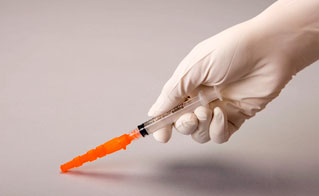
Figure 2
- Vérifier visuellement que l'aiguille est entièrement engagée dans le capuchon protecteur (figure 3). Ne retirer le dispositif Needle-Pro de la seringue avec l'aiguille fixée que si cela est requis par une procédure médicale spécifique. Enlever en saisissant le centre du dispositif de protection de l'aiguille avec le pouce et l'index, en gardant les doigts libres éloignés de l'extrémité du dispositif contenant la pointe de l'aiguille.
Figure 3
- Taper le flacon de façon ferme et répétée sur une surface dure jusqu'à ce qu'il n'y ait plus de poudre visible. Protéger la surface pour atténuer les impacts (voir figure A).

Figure A : Taper le flacon fermement pour mélanger
- Vérifier visuellement le flacon pour rechercher des grumeaux. La poudre qui n'est pas en suspension apparaît jaune, les grumeaux secs accrochent au flacon. Il peut être nécessaire de taper à nouveau le flacon si des grumeaux persistent (voir figure B).

Pas de suspension : grumeaux visibles
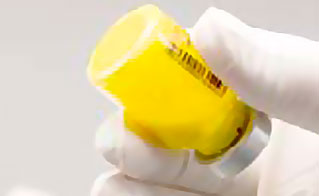
Suspension : pas de grumeaux
Figure B : Vérifier si de la poudre n'est pas en suspension et taper à nouveau si nécessaire.
- Secouer vigoureusement le flacon jusqu'à ce que la suspension apparaisse homogène et conforme au niveau de la couleur et de la texture. Le produit en suspension devient jaune et opaque (voir figure C).
Si de la mousse apparaît, laisser reposer le flacon pour qu'elle disparaisse. Si le produit n'est pas utilisé immédiatement, il devra être secoué vigoureusement pour être remis en suspension. Après reconstitution, Zypadhera reste stable jusqu'à 24 h dans le flacon.
Figure C : Secouer vigoureusement le flacon
- Tapoter légèrement le flacon pour « aérer » la poudre.
- Administration :
-
- Injection de Zypadhera :
- Le tableau ci-dessous indique les volumes finaux de suspension de Zypadhera à injecter. La concentration de la suspension est de 150 mg/ml d'olanzapine.
-
Dose (mg) Volume final à injecter (ml) 150 1,0 210 1,4 300 2,0 405 2,7 -
- Choisir quelle aiguille doit être utilisée pour administrer l'injection au patient. L'aiguille de 50 mm est recommandée pour les patients obèses :
- Si l'aiguille de 50 mm doit être utilisée pour l'injection, fixer l'aiguille sécurisée de 38 mm à la seringue pour prélever le volume de suspension nécessaire.
- Si l'aiguille de 38 mm doit être utilisée pour l'injection, fixer l'aiguille sécurisée de 50 mm à la seringue pour prélever le volume de suspension nécessaire.
- Si l'aiguille de 50 mm doit être utilisée pour l'injection, fixer l'aiguille sécurisée de 38 mm à la seringue pour prélever le volume de suspension nécessaire.
- Prélever lentement le volume désiré. Un excédent de produit restera dans le flacon.
- Fixer le dispositif de sécurité de l'aiguille et retirer l'aiguille de la seringue.
- Fixer l'aiguille sécurisée restante sur la seringue avant l'injection. Une fois que la suspension a été aspirée du flacon, elle doit être injectée immédiatement.
- Choisir et préparer un site d'injection dans le muscle fessier (glutéal). Ne pas injecter en intraveineuse ou en sous-cutané.
- Après insertion de l'aiguille, aspirer quelques secondes pour s'assurer qu'il n'y a pas de sang. Si du sang est prélevé dans la seringue, jeter la seringue et la dose préparée et recommencer la procédure de reconstitution et d'administration. L'injection doit être réalisée avec une pression constante et continue.
Ne pas masser le site d'injection.
- Fixer le dispositif de sécurité de l'aiguille (figures 1 et 2).
- Jeter les flacons, la seringue, les aiguilles et tout solvant inutilisé conformément aux procédures appropriées. Le flacon est à usage unique.
- Choisir quelle aiguille doit être utilisée pour administrer l'injection au patient. L'aiguille de 50 mm est recommandée pour les patients obèses :
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| Médicament réservé à l'usage hospitalier. | |
| Prescription réservée aux spécialistes en psychiatrie. | |
| L'administration est réservée aux services d'hospitalisation spécialisés en psychiatrie, ce qui exclut les centres médicopsychologiques et les centres d'accueil thérapeutique à temps partiel. | |
| AMM | EU/1/08/479/001 ; CIP 3400957469160 (RCP rév 01.09.2010) pdre + solv 210 mg. |
| EU/1/08/479/002 ; CIP 3400957469221 (RCP rév 01.09.2010) pdre + solv 300 mg. | |
| EU/1/08/479/003 ; CIP 3400957469399 (RCP rév 01.09.2010) pdre + solv 405 mg. | |
| Collect. |
Titulaire de l'AMM : Eli Lilly Nederland BV, Grootslag 1-5, NL-3991 RA, Houten, Pays-Bas.
LILLY FRANCE SAS
13, rue Pagès. 92158 Suresnes cdx
Standard : Tél : 01 55 49 34 34
Info médic/Pharmacovigilance :
Tél : 01 55 49 32 51 ou N° Vert : 08 00 00 36 36
Fax : 01 55 49 33 07
Logistique produits :
Tél : 01 55 49 33 21. Fax : 01 55 49 34 85
Site web : http://www.lilly.fr
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale
