BINOCRIT®
époétine alfa
FORMES et PRÉSENTATIONS |
Solution injectable à 2000 UI/1 ml (limpide ; incolore) : Seringues préremplies de 1 ml, boîtes de 1 et de 6.
Solution injectable à 3000 UI/0,3 ml (limpide ; incolore) : Seringues préremplies de 0,3 ml, boîtes de 1 et de 6.
Solution injectable à 4000 UI/0,4 ml (limpide ; incolore) : Seringues préremplies de 0,4 ml, boîtes de 1 et de 6.
Solution injectable à 5000 UI/0,5 ml (limpide ; incolore) : Seringues préremplies de 0,5 ml, boîtes de 1 et de 6.
Solution injectable à 6000 UI/0,6 ml (limpide ; incolore) : Seringues préremplies de 0,6 ml, boîtes de 1 et de 6.
Solution injectable à 8000 UI/0,8 ml (limpide ; incolore) : Seringues préremplies de 0,8 ml, boîtes de 1 et de 6.
Solution injectable à 10 000 UI/1 ml (limpide ; incolore) : Seringues préremplies de 1 ml, boîtes de 1 et de 6.
Solution injectable à 20 000 UI/0,5 ml (limpide ; incolore) : Seringue préremplie de 0,5 ml, boîte unitaire.
Solution injectable à 30 000 UI/0,75 ml (limpide ; incolore) : Seringue préremplie de 0,75 ml, boîte unitaire.
Solution injectable à 40 000 UI/1 ml (limpide ; incolore) : Seringue préremplie de 1 ml, boîte unitaire.
COMPOSITION |
| p seringue | |
| Époétine alfa* (DCI) : | |
|
- 1000 UI/0,5 ml | 8,4 µg |
|
- 2000 UI/1 ml | 16,8 µg |
|
- 3000 UI/0,3 ml | 25,2 µg |
|
- 4000 UI/0,4 ml | 33,6 µg |
|
- 5000 UI/0,5 ml | 42,0 µg |
|
- 6000 UI/0,6 ml | 50,4 µg |
|
- 8000 UI/0,8 ml | 67,2 µg |
|
- 10 000 UI/1 ml | 84 µg |
|
- 20 000 UI/0,5 ml | 168 µg |
|
- 30 000 UI/0,75 ml | 252 µg |
|
- 40 000 UI/1 ml | 336 µg |
Solution à 1000 UI/0,5 ml et 2000 UI/1 ml : chaque ml de solution contient 2000 UI d'époétine alfa*, soit 16,8 µg/ml.
Solution à 3000 UI/0,3 ml, 4000 UI/0,4 ml, 5000 UI/0,5 ml, 6000 UI/0,6 ml, 8000 UI/0,8 ml, 10 000 UI/1 ml : chaque ml de solution contient 10 000 UI d'époétine alfa*, soit 84,0 µg/ml.
Solution à 20 000 UI/0,5 ml, 30 000 UI/0,75 ml et 40 000 UI/1 ml : chaque ml de solution contient 40 000 UI d'époétine alfa*, soit 336 µg/ml.
* Produite dans des cellules d'ovaires de hamster chinois par la technique de l'ADN recombinant.
INDICATIONS |
- Traitement de l'anémie symptomatique associée à l'insuffisance rénale chronique (IRC) chez l'adulte et l'enfant :
- Traitement de l'anémie secondaire à une insuffisance rénale chronique chez les enfants et les patients adultes hémodialysés et les patients adultes en dialyse péritonéale (cf Mises en garde et Précautions d'emploi).
- Traitement de l'anémie sévère d'origine rénale accompagnée de symptômes cliniques chez les patients adultes insuffisants rénaux non encore dialysés (cf Mises en garde et Précautions d'emploi).
- Traitement de l'anémie secondaire à une insuffisance rénale chronique chez les enfants et les patients adultes hémodialysés et les patients adultes en dialyse péritonéale (cf Mises en garde et Précautions d'emploi).
- Traitement de l'anémie et réduction des besoins transfusionnels chez les patients adultes traités par chimiothérapie pour des tumeurs solides, des lymphomes malins ou des myélomes multiples et à risque de transfusion en raison de leur état général (par exemple, état cardiovasculaire, anémie préexistante au début de la chimiothérapie).
- Binocrit peut être utilisé pour augmenter les dons de sang autologue chez des malades participant à un programme de transfusions autologues différées. S'il est utilisé dans cette indication, les bénéfices doivent être évalués au regard des risques d'événements thromboemboliques signalés. Le traitement doit être administré exclusivement chez les patients présentant une anémie modérée sans carence en fer (taux d'hémoglobine [Hb] de 10-13 g/dl [6,2-8,1 mmol/l]) si les procédures d'épargne sanguine ne sont pas disponibles ou pas suffisantes lorsque l'intervention majeure non urgente prévue nécessite un volume important de sang (4 unités sanguines ou plus chez la femme, 5 unités ou plus chez l'homme).
- Binocrit peut être utilisé pour réduire l'exposition aux transfusions de sang homologue chez les patients adultes, sans carence martiale, devant subir une intervention chirurgicale orthopédique majeure programmée et présentant un risque présumé important de complications transfusionnelles. L'utilisation devra être réservée aux patients ayant une anémie modérée (par exemple, Hb de 10-13 g/dl ou 6,2-8,1 mmol/l) qui n'ont pas accès à un programme de prélèvement autologue différé et chez lesquels on s'attend à des pertes de sang de 900 à 1800 ml.
POSOLOGIE ET MODE D'ADMINISTRATION |
Le traitement par Binocrit doit être commencé sous la surveillance de médecins ayant l'expérience de la prise en charge des patients présentant les indications ci-dessus.
- Traitement de l'anémie symptomatique chez l'adulte et l'enfant en insuffisance rénale chronique :
- Chez les patients en insuffisance rénale chronique, le médicament doit être administré par voie intraveineuse (cf Mises en garde et Précautions d'emploi).
- Les symptômes et séquelles de l'anémie peuvent varier selon l'âge, le sexe et l'impact global de la maladie ; une évaluation par le médecin de l'état de santé et de l'évolution clinique de chaque patient est nécessaire.
- Le taux d'hémoglobine cible est de 10 à 12 g/dl (6,2-7,5 mmol/l) chez les adultes et de 9,5 et 11 g/dl (5,9-6,8 mmol/l) chez les enfants.
- Tout taux d'hémoglobine durablement supérieur à 12 g/dl (7,5 mmol/l) doit être évité. Si le taux d'hémoglobine connaissait une élévation supérieure à 2 g/dl (1,25 mmol/l) par mois ou s'il dépassait durablement 12 g/dl (7,5 mmol/l), il conviendrait de réduire la dose d'époétine alfa de 25 %. Si le taux d'hémoglobine dépasse 13 g/dl (8,1 mmol/l), le traitement doit être interrompu jusqu'à ce que ce taux redescende en dessous de 12 g/dl (7,5 mmol/l), après quoi le traitement par l'époétine alfa pourra être réinstauré à une dose de 25 % inférieure.
- En raison de la variabilité intra-patient, il peut arriver occasionnellement que des taux d'hémoglobine supérieurs ou inférieurs au taux souhaité soient observés.
- Les patients doivent faire l'objet d'une surveillance étroite afin de s'assurer que la plus faible dose d'époétine alfa possible soit utilisée et permette de contrôler convenablement l'anémie et les symptômes de l'anémie.
- L'état des réserves en fer devra être évalué avant et pendant le traitement et un apport complémentaire en fer devra être administré en cas de besoin. Par ailleurs, avant de commencer le traitement par l'époétine alfa, il convient d'éliminer les autres causes d'anémies, telles qu'un déficit en vitamine B12 ou en acide folique. L'absence de réponse au traitement par l'époétine alfa doit faire rechercher les causes. Celles-ci peuvent être : carence en fer, acide folique ou vitamine B12 ; intoxication par l'aluminium ; infections intercurrentes ; syndromes inflammatoires ou traumatismes ; saignements occultes ; hémolyse et fibrose médullaire de quelque origine qu'elles soient.
-
- Patients adultes en hémodialyse :
- Le traitement se déroule en deux phases :
- Phase correctrice : 50 UI/kg 3 fois par semaine par voie intraveineuse. Si un ajustement de la dose est nécessaire, il est recommandé de procéder par paliers d'au moins 4 semaines. A chaque palier, l'augmentation ou la diminution de dose préconisée est de 25 UI/kg 3 fois par semaine.
- Phase d'entretien :
La posologie est ajustée pour maintenir l'hémoglobine au taux désiré : Hb comprise entre 10 et 12 g/dl (6,2-7,5 mmol/l).
La dose hebdomadaire totale recommandée est comprise entre 75 et 300 UI/kg, administrée en doses de 25-100 UI/kg 3 fois par semaine par voie intraveineuse. Les données cliniques disponibles semblent indiquer que les patients dont le taux d'hémoglobine initial est très faible (< 6 g/dl ou < 3,75 mmol/l) pourraient avoir besoin de doses d'entretien plus importantes que ceux dont l'anémie initiale est moins sévère (Hb > 8 g/dl ou > 5 mmol/l).
- Phase correctrice : 50 UI/kg 3 fois par semaine par voie intraveineuse. Si un ajustement de la dose est nécessaire, il est recommandé de procéder par paliers d'au moins 4 semaines. A chaque palier, l'augmentation ou la diminution de dose préconisée est de 25 UI/kg 3 fois par semaine.
-
- Enfants en hémodialyse :
- Le traitement se déroule en deux phases :
- Phase correctrice :
50 UI/kg 3 fois par semaine par voie intraveineuse. Si un ajustement de la dose est nécessaire, il est recommandé de procéder par paliers de 25 UI/kg 3 fois par semaine en respectant un intervalle d'au moins 4 semaines entre chaque ajustement, jusqu'à atteindre le but désiré.
- Phase d'entretien :
La posologie est ajustée de façon à maintenir l'hémoglobine au taux désiré : Hb entre 9,5 et 11 g/dl (5,9-6,8 mmol/l).
Généralement, les enfants de moins de 30 kg nécessitent des doses d'entretien plus importantes que les enfants de plus de 30 kg et que les adultes.
A titre d'exemple, les doses d'entretien suivantes ont été utilisées lors d'essais cliniques, après 6 mois de traitement :
Les données cliniques disponibles suggèrent que les enfants dont le taux d'hémoglobine initial est très faible (< 6,8 g/dl ou < 4,25 mmol/l) peuvent avoir besoin de doses d'entretien plus importantes que ceux dont l'anémie initiale est moins sévère (Hb > 6,8 g/dl ou > 4,25 mmol/l).Dose (UI/kg 3 fois par semaine) Poids (kg) Dose médiane Dose d'entretien habituelle < 10 100 75-150 10-30 75 60-150 > 30 33 30-100
- Phase correctrice :
-
- Patients adultes en dialyse péritonéale :
- Le traitement se déroule en deux phases :
- Phase correctrice : la posologie initiale est de 50 UI/kg 2 fois par semaine par voie intraveineuse.
- Phase d'entretien : la posologie est ajustée de façon à maintenir l'hémoglobine au taux désiré : Hb entre 10 et 12 g/dl (6,2-7,5 mmol/l). La dose d'entretien est comprise entre 25 et 50 UI/kg 2 fois par semaine en 2 injections égales.
- Phase correctrice : la posologie initiale est de 50 UI/kg 2 fois par semaine par voie intraveineuse.
-
- Patients adultes insuffisants rénaux non encore dialysés :
- Le traitement se déroule en deux phases :
- Phase correctrice : la posologie initiale est de 50 UI/kg 3 fois par semaine par voie intraveineuse, suivie si nécessaire d'une augmentation de la dose de 25 UI/kg (3 fois par semaine) jusqu'à atteindre le but désiré (par palier d'au moins 4 semaines).
- Phase d'entretien : la posologie est ajustée de façon à maintenir l'hémoglobine au taux désiré : Hb comprise entre 10 et 12 g/dl (6,2-7,5 mmol/l). La dose d'entretien est comprise entre 17 et 33 UI/kg 3 fois par semaine par voie intraveineuse.
La posologie maximale ne doit pas excéder 200 UI/kg 3 fois par semaine.
- Phase correctrice : la posologie initiale est de 50 UI/kg 3 fois par semaine par voie intraveineuse, suivie si nécessaire d'une augmentation de la dose de 25 UI/kg (3 fois par semaine) jusqu'à atteindre le but désiré (par palier d'au moins 4 semaines).
- Traitement de l'anémie induite par la chimiothérapie :
- L'époétine alfa doit être administrée par voie sous-cutanée aux patients atteints d'anémie (taux d'hémoglobine <= 10 g/dl [6,2 mmol/l], par ex). Les symptômes et séquelles de l'anémie peuvent varier selon l'âge, le sexe et l'impact global de la maladie ; une évaluation par le médecin de l'état de santé et de l'évolution clinique de chaque patient est nécessaire.
- En raison de la variabilité intra-patient, il peut arriver occasionnellement que des taux d'hémoglobine supérieurs ou inférieurs au taux souhaité soient observés. Cette variabilité du taux d'hémoglobine doit être gérée en ajustant la dose de façon à le maintenir dans une marge cible de 10 g/dl (6,2 mmol/l) à 12 g/dl (7,5 mmol/l). Tout taux d'hémoglobine durablement supérieur à 12 g/dl (7,5 mmol/l) doit être évité ; les recommandations de conduite à tenir en cas de taux d'hémoglobine supérieur à 12 g/dl (7,5 mmol/l) sont décrites ci-dessous.
- Les patients doivent faire l'objet d'une surveillance étroite afin de s'assurer que la plus faible dose d'époétine alfa possible soit utilisée et permette de contrôler convenablement les symptômes de l'anémie.
- L'époétine alfa doit être administrée pendant encore un mois après la fin de la chimiothérapie.
- La dose initiale est de 150 UI/kg par voie sous-cutanée 3 fois par semaine. Alternativement, l'époétine alfa doit être administrée par voie sous-cutanée à la dose initiale de 450 UI/kg 1 fois par semaine.
- Si l'hémoglobine a augmenté d'au moins 1 g/dl (> 0,62 mmol/l) ou si les réticulocytes ont augmenté d'au mois 40 000 cellules/µl par rapport aux valeurs initiales, après 4 semaines de traitement, la dose doit être maintenue à 150 UI/kg 3 fois par semaine ou 450 UI/kg 1 fois par semaine.
- Si l'augmentation de l'hémoglobine est inférieur à 1 g/dl (< 0,62 mmol/l) et si les réticulocytes ont augmenté de moins de 40 000 cellules/µl par rapport aux valeurs initiales, la dose doit être portée à 300 UI/kg 3 fois par semaine. Si après 4 semaines supplémentaires de traitement à 300 UI/kg 3 fois par semaine, l'hémoglobine a augmenté d'au moins 1 g/dl (>= 0,62 mmol/l) ou si les réticulocytes ont augmenté d'au moins 40 000 cellules/µl, la dose de 300 UI/kg 3 fois par semaine doit être maintenue. Cependant, si l'hémoglobine a augmenté de < 1 g/dl (< 0,62 mmol/l) et si les réticulocytes ont augmenté de moins de 40 000 cellules/µl par rapport aux valeurs initiales, la réponse au traitement par l'époétine alfa est improbable et le traitement doit être interrompu.
- Si l'hémoglobine a augmenté d'au moins 1 g/dl (> 0,62 mmol/l) ou si les réticulocytes ont augmenté d'au mois 40 000 cellules/µl par rapport aux valeurs initiales, après 4 semaines de traitement, la dose doit être maintenue à 150 UI/kg 3 fois par semaine ou 450 UI/kg 1 fois par semaine.
- Le schéma posologique recommandé est décrit ci-après :
-
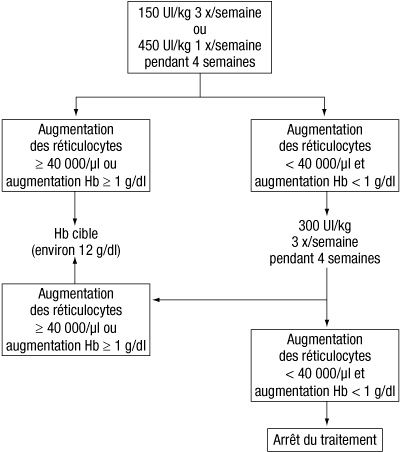
- Ajustement posologique pour maintenir le taux d'hémoglobine entre 10 g/dl et 12 g/dl (6,2-7,5 mmol/l) :
- Si le taux d'hémoglobine augmente de plus de 2 g/dl (1,25 mmol/l) par mois ou si le taux d'hémoglobine dépasse 12 g/dl (7,5 mmol/l), la dose doit être réduite d'environ 25 à 50 %. Si le taux d'hémoglobine dépasse 13 g/dl (8,1 mmol/l), le traitement doit être interrompu jusqu'à ce que ce taux redescende en dessous de 12 g/dl (7,5 mmol/l) après quoi le traitement par l'époétine alfa pourra être réinstauré à une dose de 25 % inférieure.
- Transfusion autologue programmée chez les patients adultes devant subir une intervention chirurgicale :
- Binocrit doit être administré par voie intraveineuse.
- Au moment du don de sang, Binocrit doit être administré une fois la procédure de don terminée.
- Les patients légèrement anémiques (hématocrite de 33-39 %) nécessitant un prélèvement préalable de >= 4 unités sanguines doivent être traités par Binocrit à la dose de 600 UI/kg de masse corporelle 2 fois par semaine pendant 3 semaines avant l'intervention chirurgicale.
- Tous les patients traités par Binocrit doivent recevoir une supplémentation en fer adéquat (par exemple 200 mg de fer élémentaire par voie orale chaque jour) pendant toute la durée du traitement. L'administration du complément en fer doit être instaurée dès que possible, et même plusieurs semaines avant d'entamer le prélèvement du sang autologue, afin d'atteindre des niveaux élevés de réserves en fer avant le début du traitement par Binocrit.
- Traitement des patients adultes devant bénéficier d'une intervention chirurgicale orthopédique majeure programmée :
- La voie sous-cutanée doit être utilisée.
- La dose recommandée est de 600 UI/kg d'époétine alfa, une fois par semaine pendant les 3 semaines précédant l'intervention chirurgicale (jours J-21, J-14 et J-7), ainsi que le jour de l'intervention (jour J). Dans le cas où le délai avant l'intervention doit être réduit pour des raisons médicales à moins de 3 semaines, l'époétine alfa doit être administrée quotidiennement à la dose de 300 UI/kg pendant 10 jours consécutifs avant l'intervention, ainsi que le jour de l'intervention et pendant les 4 jours suivant l'intervention. Lors du bilan hématologique préopératoire, si le taux d'hémoglobine atteint 15 g/dl (9,38 mmol/l) ou plus, l'administration d'époétine alfa doit être interrompue et les doses ultérieures initialement prévues ne doivent pas être administrées.
- Il convient de s'assurer que les patients ne présentent pas de carences en fer à l'instauration du traitement.
- Tous les patients traités par l'époétine alfa doivent recevoir un complément de fer adapté (par exemple, 200 mg/jour de Fe2+ per os) pendant toute la durée du traitement par l'époétine alfa. Si possible, la prise du complément de fer sera commencée avant le traitement par l'époétine alfa, de façon à constituer des réserves en fer suffisantes.
Mode d'administration :
Binocrit est un produit stérile, mais sans conservateur, destiné strictement à un usage unique.
Administrer la quantité requise. Ce médicament ne doit pas être administré en perfusion intraveineuse, ni en mélange avec d'autres médicaments.
- Injection par voie intraveineuse : en 1 à 5 minutes au moins, selon la dose totale. Chez les patients hémodialysés, une injection en bolus peut être réalisée pendant la séance de dialyse au site d'injection veineuse approprié de la ligne de dialyse. Une autre possibilité consiste à pratiquer l'injection à la suite de la dialyse dans la tubulure de l'aiguille à fistule, en la faisant suivre d'une injection de 10 ml de soluté isotonique afin de rincer la tubulure et d'assurer le passage correct du produit dans la circulation. Une injection plus lente est préférable chez les patients qui réagissent au traitement par des symptômes pseudogrippaux.
- Injection par voie sous-cutanée : un volume maximal de 1 ml par site d'injection ne doit pas être dépassé de façon générale. En cas de volume plus important, utiliser plusieurs sites d'injection. Les injections se font au niveau des cuisses ou de la paroi abdominale antérieure.
CONTRE-INDICATIONS |
- Hypersensibilité à la substance active ou à l'un des excipients.
- Les patients ayant développé une érythroblastopénie à la suite d'un traitement par une érythropoïétine ne doivent pas être traités par Binocrit ou par toute autre érythropoïétine (cf Mises en garde et Précautions d'emploi : Érythroblastopénies).
- Hypertension non contrôlée.
- Patients qui, quelle qu'en soit la raison, ne peuvent pas recevoir une prophylaxie antithrombotique appropriée.
- chez les patients ayant connu un infarctus du myocarde ou un AVC pendant le mois précédant le traitement ;
- ou en cas d'angor instable ;
- ou en cas de risque accru de thrombose veineuse profonde, par exemple en cas d'antécédents d'affection thromboembolique veineuse.
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
- Généralités :
- Chez tous les patients traités par époétine alfa, la tension artérielle doit être surveillée étroitement et contrôlée de façon appropriée. L'époétine alfa doit être utilisée avec précaution en présence d'une hypertension non ou insuffisamment traitée ou difficilement contrôlable. Il peut être nécessaire d'instaurer ou d'augmenter le traitement antihypertenseur. Si la pression artérielle ne peut être contrôlée, le traitement par époétine alfa doit être interrompu.
- L'époétine alfa doit également être utilisée avec précaution en présence d'épilepsie ou d'insuffisance hépatique chronique.
- Chez les patients atteints d'insuffisance rénale chronique et de cancer traités par l'époétine alfa, les taux d'hémoglobine doivent être mesurés régulièrement jusqu'à ce qu'ils se stabilisent, et de façon périodique par la suite.
- Les taux d'hémoglobine doivent être étroitement surveillés chez l'ensemble des patients en raison du risque potentiellement accru d'accidents thromboemboliques et d'issue fatale lorsque le traitement est administré en présence de taux d'hémoglobine supérieurs à la valeur cible définie pour l'indication.
- Lors d'un traitement par époétine alfa, il est possible d'observer une augmentation de la numération plaquettaire modérée dose-dépendante, dans les limites de la normale. Celle-ci régresse avec la poursuite du traitement. Il est conseillé de surveiller la numération plaquettaire à intervalles réguliers pendant les 8 premières semaines de traitement.
- Toutes les autres causes d'anémie (carence en fer, hémolyse, pertes sanguines, déficit en vitamine B12 ou en acide folique) doivent être prises en compte et traitées avant d'initier le traitement par époétine alfa. Dans la plupart des cas, les taux de ferritine sérique chutent parallèlement à l'augmentation de l'hématocrite. Afin d'obtenir une réponse optimale au traitement par époétine alfa, il convient de s'assurer que les réserves en fer sont suffisantes :
- L'administration d'un complément de fer est recommandée chez les insuffisants rénaux chroniques ayant des taux de ferritine sérique inférieurs à 100 ng/ml, sur la base de 200-300 mg/jour de Fe2+ per os (100-200 mg/jour chez l'enfant).
- Un traitement substitutif en Fe2+ de 200 à 300 mg/jour per os est recommandé chez tous les patients cancéreux dont le coefficient de saturation de transferrine est inférieur à 20 %.
- L'administration d'un complément de fer est recommandée chez les insuffisants rénaux chroniques ayant des taux de ferritine sérique inférieurs à 100 ng/ml, sur la base de 200-300 mg/jour de Fe2+ per os (100-200 mg/jour chez l'enfant).
- Chez les patients cancéreux, tous ces autres facteurs d'anémie mentionnés doivent également être soigneusement examinés avant de décider d'augmenter la posologie de l'époétine alfa.
- Les bonnes pratiques de gestion du sang doivent toujours être appliquées dans le contexte chirurgical.
- Afin d'améliorer la traçabilité des agents stimulant l'érythropoïèse (ESA), le nom de l'ESA administré doit être clairement précisé dans le dossier du patient.
- Érythroblastopénies :
- Des érythroblastopénies avec anticorps ont été rapportées dans de très rares cas après plusieurs mois ou années de traitement par époétine par voie sous-cutanée. Chez les patients présentant une perte soudaine d'efficacité définie par une baisse de l'hémoglobine (de 1 à 2 g/dl par mois ou 0,62 à 1,25 mmol/l par mois), avec augmentation des besoins transfusionnels, une numération des réticulocytes devra être réalisée et les causes habituelles de non-réponse (par exemple carence en fer, acide folique ou vitamine B12, intoxication à l'aluminium, infection ou inflammation, pertes sanguines et hémolyse) devront être recherchées.
- Si le taux de réticulocytes corrigé en fonction de l'anémie (c'est-à-dire l'index réticulocytaire) est faible (< 20 000/mm3 ou < 20 000/µl ou < 0,5 %), les plaquettes et les leucocytes étant normaux, et si aucune autre cause de perte d'efficacité n'a été trouvée, des anticorps anti-érythropoïétine devront être recherchés et une ponction médullaire devra être envisagée pour confirmer le diagnostic d'érythroblastopénie.
- Si une érythroblastopénie avec anticorps anti-érythropoïétine est suspectée, le traitement par Binocrit doit être immédiatement interrompu. Aucun traitement par une autre érythropoïétine ne devra être débuté en raison du risque de réaction croisée. Un traitement adapté, tel que des transfusions sanguines, pourra être envisagé au besoin.
- L'apparition d'une diminution paradoxale du taux d'hémoglobine et d'une anémie sévère associée à de faibles numérations des réticulocytes impose d'arrêter le traitement par l'époétine et de rechercher des anticorps anti-érythropoïétine. De tels cas ont été signalés chez des patients atteints d'hépatite C traités par interféron et ribavirine, lors de l'utilisation concomitante d'époétines. Les époétines n'ont pas été approuvées dans le cadre de la prise en charge de l'anémie associée à l'hépatite C.
- Patients en insuffisance rénale chronique :
- Les données concernant l'immunogénicité en cas d'administration par voie sous-cutanée de Binocrit chez les patients à risque d'érythroblastopénie avec anticorps, c'est-à-dire les patients atteints d'anémie rénale, sont insuffisantes. Le médicament doit donc être administré par voie intraveineuse chez ces patients.
- Chez les patients en insuffisance rénale chronique, le taux d'hémoglobine doit augmenter d'environ 1 g/dl (0,62 mmol/l) par mois et ne pas dépasser 2 g/dl (1,25 mmol/l) par mois afin de limiter au maximum les risques d'aggravation d'une hypertension.
- Chez les patients en insuffisance rénale chronique, le taux d'hémoglobine à la dose d'entretien ne doit pas dépasser la limite supérieure du taux d'hémoglobine cible recommandée dans la rubrique Posologie/Mode d'administration. Lors des essais cliniques, un risque accru de décès et d'événements cardiovasculaires graves a été observé lorsque des agents stimulant l'érythropoïèse (ESA) ont été administrés avec pour cible un taux d'hémoglobine supérieur à 12 g/dl (7,5 mmol/l).
- Les essais cliniques contrôlés n'ont pas montré d'effets bénéfiques significatifs attribuables à l'administration des époétines lorsque le taux d'hémoglobine dépassait le niveau nécessaire au contrôle des symptômes de l'anémie et pour éviter une transfusion sanguine.
- Des thromboses du shunt se sont produites chez des patients sous hémodialyse, en particulier chez les patients ayant tendance à l'hypotension ou présentant des complications au niveau de leur fistule artérioveineuse (par ex., sténoses, anévrismes, etc.). Une révision anticipée du shunt et une prophylaxie antithrombotique par administration d'acide acétylsalicylique, par exemple, est recommandée chez ces patients.
- Une hyperkaliémie a été observée dans des cas isolés. La correction de l'anémie peut entraîner une augmentation de l'appétit et de l'apport en potassium et en protéine. Il peut s'avérer nécessaire d'ajuster périodiquement les modalités de prescription de la dialyse pour maintenir l'urée, la créatinine et le potassium dans les limites désirées. L'ionogramme sanguin doit être contrôlé chez les insuffisants rénaux chroniques. En cas d'hyperkaliémie (ou d'augmentation de la kaliémie), l'arrêt de l'administration d'époétine alfa jusqu'à correction de l'hyperkaliémie doit être envisagé.
- Lors d'un traitement par époétine alfa, l'augmentation de l'hématocrite rend souvent nécessaire d'augmenter les doses d'héparine pendant l'hémodialyse. Une obstruction du système de dialyse peut survenir si l'héparinisation n'est pas optimale.
- D'après les données disponibles à ce jour, la correction de l'anémie par époétine alfa chez les patients insuffisants rénaux non encore dialysés n'accélère pas l'évolution de l'insuffisance rénale.
- Patients cancéreux adultes présentant une anémie symptomatique et recevant une chimiothérapie :
- Les érythropoïétines sont des facteurs de croissance qui stimulent essentiellement la production des globules rouges. Des récepteurs à l'érythropoïétine peuvent être exprimés à la surface de diverses cellules malignes. Comme pour tout facteur de croissance, il n'est pas exclu que les époétines puissent stimuler la croissance des tumeurs. Lors de plusieurs essais contrôlés, les époétines n'ont pas fait la preuve de leur capacité à améliorer le taux de survie globale ou à réduire le risque de progression tumorale chez les patients atteints d'anémie associée au cancer.
- Lors des essais cliniques contrôlés de l'époétine alfa et d'autres agents stimulant l'érythropoïèse, les résultats suivants ont été mis en évidence :
- réduction du contrôle locorégional chez des patients atteints de cancers de la tête et du cou traités par radiothérapie lorsque l'administration visait un taux d'hémoglobine supérieur à 14 g/dl (8,7 mmol/l) ;
- raccourcissement de la durée de survie globale et augmentation des décès imputables à la progression de la maladie à 4 mois chez des patients atteints de cancers du sein métastatiques traités par chimiothérapie lorsque l'administration visait un taux d'hémoglobine de 12 à 14 g/dl (7,5-8,7 mmol/ml) ;
- augmentation du risque de décès lorsque l'administration visait un taux d'hémoglobine de 12 g/dl (7,5 mmol/l) chez des patients atteints de tumeurs actives et ne recevant ni chimiothérapie ni radiothérapie. L'utilisation des ESA n'est pas indiquée chez cette population de patients.
- réduction du contrôle locorégional chez des patients atteints de cancers de la tête et du cou traités par radiothérapie lorsque l'administration visait un taux d'hémoglobine supérieur à 14 g/dl (8,7 mmol/l) ;
- Au vu des informations ci-dessus, dans certaines situations cliniques, la transfusion sanguine doit être le traitement privilégié de l'anémie des patients cancéreux. La décision d'administrer des érythropoïétines recombinantes doit être déterminée sur la base d'une évaluation du rapport bénéfice/risque prenant en compte l'avis du patient dans son contexte clinique spécifique. Les facteurs à considérer dans cette évaluation doivent inclure le type de tumeur et son stade, le degré de l'anémie, l'espérance de vie, l'environnement dans lequel le patient est traité et la préférence du patient (cf Pharmacodynamie).
- Lors de l'évaluation du caractère approprié d'un traitement par époétine alfa chez les patients cancéreux traités par chimiothérapie (patients à risque d'être transfusés), il faut tenir compte du fait que l'apparition des globules rouges suit l'administration de l'érythropoïétine avec un délai de 2 à 3 semaines.
- Afin de limiter au maximum les risques de survenue d'événements thrombotiques, le taux d'hémoglobine et son augmentation ne doivent pas dépasser les limites indiquées dans la rubrique Posologie et Mode d'administration.
- Une augmentation de l'incidence des événements vasculaires thrombotiques (EVT) ayant été observée chez les patients cancéreux recevant des agents stimulant l'érythropoïèse (cf Effets indésirables), ce risque doit être soigneusement évalué au regard du bénéfice de ce traitement (par époétine alfa), particulièrement chez les patients cancéreux présentant un risque accru d'événements vasculaires thrombotiques, comme en cas d'obésité ou d'antécédents d'EVT (par exemple, de thrombose veineuse profonde ou d'embolie pulmonaire). Une étude de recherche (étude BEST) menée chez des femmes atteintes de cancers du sein métastatiques a été mise au point pour déterminer si le traitement par l'époétine alfa allant au-delà de la correction de l'anémie pouvait améliorer l'issue du traitement. Lors de cette étude, l'incidence des accidents thromboemboliques fatals a été plus élevée chez les patientes sous époétine alfa que chez celles recevant le placebo (cf Pharmacodynamie).
- Transfusion autologue programmée chez les patients adultes devant subir une intervention chirurgicale :
- Toutes les mises en garde et précautions d'emploi particulières associées aux transfusions autologues programmées, en particulier celles liées au remplissage vasculaire de routine, doivent être respectées.
- Patients adultes devant bénéficier d'une intervention chirurgicale orthopédique majeure programmée :
- Chez les patients devant bénéficier d'une intervention chirurgicale orthopédique majeure programmée, la cause de l'anémie doit être établie et traitée, si possible avant l'instauration du traitement par époétine alfa. Les événements thromboemboliques peuvent constituer un risque dans cette population, risque qui doit être soigneusement évalué au regard du bénéfice potentiel du traitement chez ces patients.
- Les patients devant bénéficier d'une intervention chirurgicale orthopédique majeure programmée doivent recevoir une prophylaxie antithrombotique appropriée, dans la mesure où des événements thromboemboliques peuvent survenir chez ces patients, particulièrement en présence d'une pathologie cardiovasculaire sous-jacente. En outre, des précautions particulières doivent être prises chez les patients à risque de développer des thromboses veineuses profondes (TVP). De plus, chez les patients dont le taux d'hémoglobine initial est supérieur à 13 g/dl (> 8,1 mmol/l), la possibilité que le traitement par époétine alfa soit associé à un risque accru d'événements thromboemboliques postopératoires ne peut être exclue. En conséquence, l'époétine alfa ne doit pas être utilisée chez les patients dont le taux d'hémoglobine initial est supérieur à 13 g/dl (> 8,1 mmol/l).
- Excipients :
- Ce médicament contient moins de 1 mmol de sodium (23 mg) par seringue préremplie, c'est-à-dire qu'il est pratiquement « exempt de sodium ».
INTERACTIONS |
Aucune donnée n'indique une éventuelle interaction de l'époétine alfa avec le métabolisme d'autres médicaments. Cependant, étant donné que la ciclosporine se lie aux hématies, une interaction demeure possible. Si l'époétine alfa est administrée en association avec la ciclosporine, les taux sanguins de ciclosporine doivent être surveillés et la dose de ciclosporine ajustée en fonction de l'augmentation de l'hématocrite.
Aucune donnée n'indique une interaction entre le facteur de croissance des granulocytes (G-CSF) ou le facteur de croissance des granulocytes et macrophages (GM-CSF) et l'époétine alfa en ce qui concerne la différenciation ou la prolifération hématologique des échantillons de biopsies tumorales in vitro.
FERTILITÉ/GROSSESSE/ALLAITEMENT |
En conséquence :
- chez l'insuffisant rénal chronique, Binocrit ne doit être utilisé en cas de grossesse que si le bénéfice escompté contrebalance le risque potentiel pour le foetus ;
- l'utilisation d'époétine alfa n'est pas recommandée en cas de grossesse ou d'allaitement chez des patientes devant subir une intervention chirurgicale avec transfusion autologue programmée.
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
- Généralités :
- Chez les patients cancéreux et les patients en insuffisance rénale chronique, la réaction indésirable la plus fréquente pendant le traitement par l'époétine alfa est une élévation de la pression artérielle en fonction de la dose ou une aggravation d'une hypertension existante. La pression artérielle doit être surveillée, en particulier en début de traitement (cf Mises en garde et Précautions d'emploi). Les autres réactions indésirables fréquentes observées dans les essais cliniques de l'époétine alfa ont été des thromboses veineuses profondes, des embolies pulmonaires, des crises, des diarrhées, des nausées, des céphalées, des symptômes pseudogrippaux, des pyrexies, des éruptions cutanées et des vomissements. Les symptômes pseudogrippaux, incluant notamment des céphalées, une arthralgie, une myalgie et une pyrexie, peuvent apparaître en particulier en début de traitement. La fréquence des réactions peut varier selon l'indication (voir le tableau ci dessous).
- Les réactions indésirables graves au médicament sont notamment des thromboses veineuses et artérielles et des embolies (fatales dans certains cas), telles que des thromboses veineuses profondes, des embolies pulmonaires, des thromboses artérielles (notamment des infarctus du myocarde et des ischémies myocardiaques), des thromboses rétiniennes et des thromboses du shunt (y compris liées au matériel de dialyse). Par ailleurs, des accidents vasculaires cérébraux (notamment des infarctus cérébraux et des hémorragies cérébrales) et des accidents ischémiques transitoires ont été rapportés lors des essais cliniques de l'époétine alfa.
- Des cas d'anévrismes ont été rapportés.
- Des réactions d'hypersensibilité, notamment des cas d'éruptions cutanées, d'urticaire, de réactions anaphylactiques et d'oedèmes de Quincke, ont été rapportées.
- Des crises hypertensives accompagnées d'encéphalopathies et de crises convulsives, nécessitant une prise en charge médicale immédiate et des soins intensifs, se sont également produites pendant le traitement par l'époétine alfa chez des patients dont la pression artérielle était auparavant normale ou faible. Les céphalées de type migraine violente et à début brutal peuvent en être le signal d'alarme et doivent faire l'objet d'une attention particulière.
- De très rares cas d'érythroblastopénie avec anticorps ont été signalés (< 1/10 000 cas par patient-année) après plusieurs mois ou années de traitement par l'époétine alfa (cf Mises en garde et Précautions d'emploi).
- Le profil de sécurité global de l'époétine alfa a été évalué chez 142 sujets atteints d'insuffisance rénale chronique et chez 765 sujets cancéreux ayant participé à des essais cliniques en double aveugle, contrôlés contre placebo, pour l'autorisation du produit. Les réactions indésirables au médicament rapportées chez >= 0,2 % des sujets de ces essais traités par l'époétine alfa, ainsi que dans les essais cliniques supplémentaires et en phase de pharmacovigilance, sont énumérées ci dessous par classe de système d'organes et par fréquence.
- Les fréquences sont définies ainsi : très fréquent (>= 1/10) ; fréquent (>= 1/100, < 1/10) ; peu fréquent (>= 1/1000, < 1/100) ; rare (>= 1/10 000, < 1/1000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
- Au sein de chaque groupe de fréquence, les effets indésirables doivent être présentés suivant un ordre décroissant de gravité.
-
Classe de système d'organes et fréquence Réactions indésirables Affections hématologiques et du système lymphatique Peu fréquent Thrombocytopénie (patients cancéreux) Fréquence indéterminée Érythroblastopénie avec anticorps anti-érythropoïétine*
Thrombocytémie (patients en insuffisance rénale chronique)Affections du système immunitaire Fréquence indéterminée Réaction anaphylactique
HypersensibilitéAffections du système nerveux Très fréquent Céphalées (patients cancéreux) Fréquent Crises convulsives (patients en insuffisance rénale chronique)
Céphalées (patients en insuffisance rénale chronique)Peu fréquent Hémorragie cérébrale**
Crises convulsives (patients cancéreux)Fréquence indéterminée Accident vasculaire cérébral**
Encéphalopathie hypertensive
Accident ischémique transitoireAffections oculaires Fréquence indéterminée Thrombose rétinienne Affections vasculaires Fréquent Thrombose veineuse profonde** (patients cancéreux)
HypertensionFréquence indéterminée Thrombose veineuse profonde** (patients en insuffisance rénale chronique)
Thrombose artérielle
Crise hypertensiveAffections respiratoires, thoraciques et médiastinales Fréquent Embolie pulmonaire** (patients cancéreux) Fréquence indéterminée Embolie pulmonaire** (patients en insuffisance rénale chronique) Affections gastro-intestinales Très fréquent Nausées Fréquent Diarrhées (patients cancéreux)
VomissementsPeu fréquent Diarrhées (patients en insuffisance rénale chronique) Affections de la peau et du tissu sous-cutané Fréquent Éruption cutanée Fréquence indéterminée OEdème de Quincke
UrticaireAffections musculosquelettiques et systémiques Très fréquent Arthralgie (patients en insuffisance rénale chronique) Fréquent Arthralgie (patients cancéreux) Peu fréquent Myalgie (patients cancéreux) Fréquence indéterminée Myalgie (patients en insuffisance rénale chronique) Affections congénitales, familiales et génétiques Fréquence indéterminée Porphyrie Troubles généraux et anomalies au site d'administration Très fréquent Pyrexie (patients cancéreux)
Symptômes pseudogrippaux (patients en insuffisance rénale chronique)Fréquent Symptômes pseudogrippaux (patients cancéreux) Fréquence indéterminée Inefficacité du médicament
OEdème périphérique
Pyrexie (patients en insuffisance rénale chronique)
Réaction au site d'injectionInvestigations Fréquence indéterminée Patient positif aux anticorps anti-érythropoïétine* Lésions, intoxications et complications liées aux procédures Fréquent Thrombose du shunt y compris du matériel de dialyse (patients en insuffisance rénale chronique) -
*
Réactions dont la fréquence n'a pu être estimée d'après les essais cliniques.
-
**
Y compris des cas dont l'issue a été fatale.
- Patients en insuffisance rénale chronique :
- Chez les patients en insuffisance rénale chronique, les taux d'hémoglobine supérieurs à 12 g/dl (7,5 mmol/l) peuvent être associés à un risque accru d'accidents cardiovasculaires, y compris de décès (cf Mises en garde et Précautions d'emploi).
- Des thromboses du shunt se sont produites chez des patients sous hémodialyse, en particulier chez les patients ayant tendance à l'hypotension ou présentant des complications au niveau de leur fistule artérioveineuse (par exemple : sténoses, anévrismes, etc.), cf Mises en garde et Précautions d'emploi.
- Patients cancéreux :
- Une augmentation de l'incidence des accidents thromboemboliques a été rapportée chez les patients cancéreux recevant des agents stimulant l'érythropoïèse (ESA), y compris l'époétine alfa (cf Mises en garde et Précautions d'emploi).
- Patients devant bénéficier d'une intervention chirurgicale :
- Chez les patients devant bénéficier d'une intervention chirurgicale orthopédique majeure programmée et présentant un taux d'hémoglobine initial de 10 à 13 g/dl (6,2-8,1 mmol/l), l'incidence des événements thromboemboliques (dont la plupart étaient des thromboses veineuses profondes) sur l'ensemble des essais cliniques, s'est avérée similaire dans les différents groupes traités par époétine alfa et dans le groupe placebo. Néanmoins, l'expérience clinique est limitée.
- Par ailleurs, chez les patients dont le taux d'hémoglobine initial est > 13 g/dl (8,1 mmol/l), la possibilité que le traitement par époétine alfa puisse être associé à une augmentation du risque d'événements thromboemboliques postopératoires ne peut être exclue.
SURDOSAGE |
PHARMACODYNAMIE |
Classe pharmacothérapeutique : autres préparations antianémiques (code ATC : B03XA01).
L'érythropoïétine est une glycoprotéine qui stimule la formation des érythrocytes à partir des cellules souches de la moelle osseuse ; elle agit à ce niveau en tant qu'hormone de différenciation et facteur stimulant les mitoses.
Le poids moléculaire apparent de l'érythropoïétine se situe entre 32 000 et 40 000 daltons. La fraction protéique représente environ 58 % de la molécule et est constituée de 165 acides aminés. Les quatre chaînes glucidiques sont rattachées à la protéine par trois liaisons N-glycosidique et une liaison O-glycosidique. L'époétine alfa obtenue par génie génétique est glycosylée. Sa composition en aminoacides et glucides est identique à celle de l'érythropoïétine endogène humaine isolée des urines de patients anémiques.
Binocrit dispose du degré de pureté le plus élevé actuellement possible. En particulier, aucun résidu de la lignée cellulaire utilisée dans sa production n'est détectable aux concentrations actives chez l'être humain.
L'efficacité biologique de l'époétine alfa a été démontrée grâce aux différents modèles in vivo chez l'animal (rats normaux et anémiques, souris ayant une polyglobulie). Après administration d'époétine alfa, la numération érythrocytaire, le taux d'hémoglobine et la numération des réticulocytes augmentent, de même que le taux d'incorporation du 59Fe.
Après incubation de cellules nucléées érythroïdes spléniques in vitro avec l'époétine alfa (culture cellulaire de cellules spléniques de souris), il a été observé une augmentation de l'incorporation de la 3H-thymidine.
Grâce à des cellules de moelle osseuse humaine en culture, il a pu être démontré que l'époétine alfa stimule spécifiquement l'érythropoïèse sans avoir d'effets sur la leucopoïèse. Aucune action cytotoxique de l'époétine alfa sur les cellules de moelle osseuse humaine n'a été détectée.
721 patients cancéreux recevant une chimiothérapie sans platine ont été inclus dans 3 études contrôlées contre placebo. Parmi eux, 389 patients présentaient des hémopathies malignes (221 myélomes multiples, 144 lymphomes non hodgkiniens et 24 autres hémopathies malignes) et 332 patients présentaient des tumeurs solides (172 cancers du sein, 64 cancers gynécologiques, 23 cancers du poumon, 22 cancers de la prostate, 21 cancers gastro-intestinaux et 30 cancers d'autres types). Dans 2 larges études ouvertes, 2697 patients cancéreux recevant une chimiothérapie sans platine ont été inclus ; 1895 patients présentaient des tumeurs solides (683 cancers du sein, 260 cancers du poumon, 174 cancers gynécologiques, 300 cancers gastro-intestinaux et 478 cancers d'autres types) et 802 présentaient des hémopathies malignes.
Dans une étude prospective, randomisée, en double aveugle, contrôlée contre placebo, conduite chez 375 patients anémiques recevant une chimiothérapie sans platine pour diverses hémopathies malignes de type non myéloïdes, il a été observé une diminution significative des conséquences de l'anémie (par exemple, fatigue, baisse d'énergie et réduction de l'activité) mesurées par les instruments et échelles suivantes : échelle générale d'évaluation fonctionnelle du traitement de l'anémie du cancer FACT-an, échelle de fatigue FACT-an et échelle analogique linéaire du cancer (CLAS). Deux autres études randomisées, contrôlées contre placebo, d'effectifs plus réduit, n'ont pas permis de montrer une amélioration significative des paramètres de qualité de vie sur les échelles EORTC-QLQ-C30 et CLAS, respectivement.
L'érythropoïétine est un facteur de croissance qui stimule essentiellement la production des globules rouges. Des récepteurs à l'érythropoïétine peuvent être exprimés à la surface de diverses cellules malignes.
La survie et la progression tumorale ont été étudiées dans le cadre de cinq vastes essais cliniques contrôlés portant au total sur 2833 patients, dont quatre essais en double aveugle contrôlés contre placebo et un essai ouvert. Les études ont recruté des patients traités par chimiothérapie (deux études) ou se sont basées sur des populations de patients chez lesquelles l'utilisation d'agents stimulant l'érythropoïèse n'est pas indiquée : anémie touchant des patients cancéreux non traités par chimiothérapie et patients atteints de cancers de la tête et du cou traités par radiothérapie. Dans deux des études, le taux d'hémoglobine cible était > 13 g/dl (8,1 mmol/l) ; dans les trois autres études, il était de 12 à 14 g/dl (7,5-8,7 mmol/l). Dans l'essai ouvert, aucune différence n'a été notée en terme de survie globale entre les patients traités par l'érythropoïétine humaine recombinante et les sujets témoins. Dans les quatre essais contrôlés contre placebo, les rapports de risque pour la survie globale ont été compris entre 1,25 et 2,47 en faveur des groupes témoins. Ces études ont fait apparaître de façon cohérente un surplus statistiquement significatif et inexpliqué de mortalité chez les patients atteints d'anémie associée à divers cancers courants et recevant une érythropoïétine humaine recombinante par comparaison avec les sujets témoins. Les différences d'incidence des thromboses et complications associées entre les sujets recevant l'érythropoïétine humaine recombinante et les sujets du groupe témoin ne suffisent pas à expliquer de façon satisfaisante les résultats des essais concernant la survie globale.
Un passage en revue systématique a également été appliqué à plus de 9000 patients cancéreux participant à 57 essais cliniques. La méta-analyse des données de survie globale a donné un rapport de risque estimé ponctuellement à 1,08 en faveur des sujets témoins (IC à 95 % : 0,99-1,18 ; 42 essais et 8167 patients). Un risque relatif accru d'événements thromboemboliques (RR = 1,67 ; IC à 95 % : 1,35-2,06 ; 35 essais et 6769 patients) a été observé chez les patients traités par une érythropoïétine humaine recombinante. Il existe un risque accru d'accident thromboembolique chez les patients cancéreux traités par une érythropoïétine humaine recombinante et l'on ne saurait exclure la possibilité d'un impact négatif sur la survie globale. Il est difficile de savoir dans quelle mesure ces résultats peuvent s'appliquer à l'administration d'une érythropoïétine humaine recombinante chez les patients cancéreux traités par chimiothérapie avec pour objectif un taux d'hémoglobine inférieur à 13 g/dl (8,1 mmol/l) car les données passées en revue incluaient peu de patients réunissant ces caractéristiques.
Une analyse des données individuelles a également été réalisée sur plus de 13 900 patients cancéreux (sous chimiothérapie, radiothérapie, chimioradiothérapie ou non traités) participant à 53 essais cliniques contrôlés portant sur plusieurs époétines. La méta-analyse des données de survie globale a donné un rapport de risque estimé ponctuellement à 1,06 en faveur des sujets témoins (IC à 95 % : 1,00, 1,12 ; 53 essais et 13 933 patients) et, chez les patients cancéreux sous chimiothérapie, le rapport de risque de la survie globale a été de 1,04 (IC à 95 % : 0,97, 1,11 ; 38 essais et 10 441 patients). Les méta-analyses indiquent également de façon cohérente un risque relatif significativement accru d'accident thromboembolique chez les patients cancéreux recevant une érythropoïétine humaine recombinante (cf Mises en garde et Précautions d'emploi).
PHARMACOCINÉTIQUE |
- Par voie intraveineuse :
- Le dosage de l'époétine alfa après administration intraveineuse répétée a montré une demi-vie de 4 heures environ chez les volontaires sains et légèrement plus longue chez les insuffisants rénaux, soit environ 5 heures. Chez l'enfant, la demi-vie est d'environ 6 heures.
- Par voie sous-cutanée :
- Après injection sous-cutanée, les taux sériques d'époétine alfa sont largement inférieurs aux taux obtenus après injection intraveineuse ; les taux augmentent lentement pour atteindre leur valeur maximale entre 12 à 18 heures après administration. Le pic est toujours très inférieur au pic obtenu par voie intraveineuse (environ 1/20e).
- Il n'y a pas d'accumulation du produit : les taux demeurent identiques, qu'ils soient mesurés 24 heures après la première injection ou 24 heures après la dernière injection.
- La demi-vie est difficile à évaluer pour la voie sous-cutanée mais elle est estimée à environ 24 heures. La biodisponibilité de l'époétine alfa injectable par voie sous-cutanée est très inférieure à celle du produit administré par voie intraveineuse : environ 20 %.
SÉCURITE PRÉCLINIQUE |
Lors de certaines études précliniques de toxicologie chez le chien et le rat, mais pas chez le singe, le traitement par époétine alfa a été associé à une fibrose infraclinique de la moelle osseuse. (La fibrose médullaire est une complication connue de l'insuffisance rénale chronique chez l'être humain et pourrait être liée à une hyperparathyroïdie secondaire ou à des facteurs encore inconnus. L'incidence de la fibrose médullaire n'a pas été augmentée lors d'une étude chez des patients sous hémodialyse traités par époétine alfa pendant 3 ans, par rapport à un groupe témoin apparié de patients sous dialyse qui n'ont pas été traités par époétine alfa.)
Chez l'animal, il a été démontré que l'époétine alfa, à une dose hebdomadaire d'environ 20 fois supérieure à la dose hebdomadaire recommandée chez l'être humain, diminuait le poids foetal, retardait l'ossification et augmentait la mortalité foetale. Ces modifications seraient secondaires à la diminution de la prise de poids chez la mère.
L'époétine alfa n'a pas montré de modification des tests de mutagénicité des cultures de cellules de bactéries et de mammifères, et in vivo dans le test du micronucleus chez la souris.
Les études de cancérogénicité à long terme n'ont pas été réalisées. Il existe des rapports contradictoires dans la littérature concernant le rôle potentiel majeur des érythropoïétines dans la prolifération tumorale. Ces rapports sont basés sur des observations in vitro portant sur des échantillons de tumeurs humaines mais leur pertinence en pratique clinique est incertaine.
INCOMPATIBILITÉS |
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 2 ans.
A conserver et transporter réfrigéré (entre 2 °C et 8 °C). Ne pas congeler.
Conserver la seringue préremplie dans l'emballage extérieur à l'abri de la lumière.
Pour l'usage ambulatoire, le patient peut sortir Binocrit du réfrigérateur et le conserver à une température ne dépassant pas 25 °C pour une période unique de 3 jours maximum.
MODALITÉS MANIPULATION/ÉLIMINATION |
- si la solution est trouble ou si elle contient des particules ;
- si le conditionnement est endommagé ;
- si la solution a été accidentellement congelée.
Les seringues préremplies sont prêtes à l'emploi (cf Posologie et Mode d'administration). La seringue préremplie ne doit pas être secouée. Des graduations sont gravées sur les seringues afin de permettre une utilisation partielle si nécessaire. Chaque graduation correspond à un volume de 0,1 ml. Prendre une dose de Binocrit uniquement dans chaque seringue, et jeter la solution restante avant l'injection.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| Médicament soumis à prescription initiale hospitalière annuelle. | |
| La prescription initiale par un médecin exerçant dans un service de dialyse à domicile est également autorisée. | |
| AMM | EU/1/07/410/001 ; CIP 3400938214536 (1 × 1000 UI). |
| EU/1/07/410/002 ; CIP 3400938214765 (6 × 1000 UI). | |
| EU/1/07/410/003 ; CIP 3400938214826 (1 × 2000 UI). | |
| EU/1/07/410/004 ; CIP 3400938214994 (6 × 2000 UI). | |
| EU/1/07/410/005 ; CIP 3400938215076 (1 × 3000 UI). | |
| EU/1/07/410/006 ; CIP 3400938215137 (6 × 3000 UI). | |
| EU/1/07/410/007 ; CIP 3400938215366 (1 × 4000 UI). | |
| EU/1/07/410/008 ; CIP 3400938215427 (6 × 4000 UI). | |
| EU/1/07/410/009 ; CIP 3400938215595 (1 × 5000 UI). | |
| EU/1/07/410/010 ; CIP 3400938215656 (6 × 5000 UI). | |
| EU/1/07/410/011 ; CIP 3400938215717 (1 × 6000 UI). | |
| EU/1/07/410/012 ; CIP 3400938215885 (6 × 6000 UI). | |
| EU/1/07/410/013 ; CIP 3400938216028 (1 × 8000 UI). | |
| EU/1/07/410/014 ; CIP 3400938216318 (6 × 8000 UI). | |
| EU/1/07/410/015 ; CIP 3400938216776 (1 × 10 000 UI). | |
| EU/1/07/410/016 ; CIP 3400938216837 (6 × 10 000 UI). | |
| EU/1/07/410/021 ; CIP 3400939900261 (2009) 1 × 20 000 UI. | |
| EU/1/07/410/023 ; CIP 3400939900490 (2009) 1 × 30 000 UI. | |
| EU/1/07/410/025 ; CIP 3400939900612 (2009) 1 × 40 000 UI. | |
| RCP révisés le 14.03.2011. | |
| Prix : | 8.49 euros (1 seringue à 1000 UI/0,5 ml). |
| 45.87 euros (6 seringues à 1000 UI/0,5 ml). | |
| 16.43 euros (1 seringue à 2000 UI/1 ml). | |
| 86.52 euros (6 seringues à 2000 UI/1 ml). | |
| 24.37 euros (1 seringue à 3000 UI/0,3 ml). | |
| 127.17 euros (6 seringues à 3000 UI/0,3 ml). | |
| 32.32 euros (1 seringue à 4000 UI/0,4 ml). | |
| 167.82 euros (6 seringues à 4000 UI/0,4 ml). | |
| 39.10 euros (1 seringue à 5000 UI/0,5 ml). | |
| 206.70 euros (6 seringues à 5000 UI/0,5 ml). | |
| 45.87 euros (1 seringue à 6000 UI/0,6 ml). | |
| 244.54 euros (6 seringues à 6000 UI/0,6 ml). | |
| 59.42 euros (1 seringue à 8000 UI/0,8 ml). | |
| 320.23 euros (6 seringues à 8000 UI/0,8 ml). | |
| 72.97 euros (1 seringue à 10 000 UI/1 ml). | |
| 395.92 euros (6 seringues à 10 000 UI/1 ml). | |
| 140.72 euros (1 seringue à 20 000 UI/0,5 ml). | |
| 206.69 euros (1 seringue à 30 000 UI/0,75 ml). | |
| 269.76 euros (1 seringue à 40 000 UI/1 ml). | |
|
Remb Séc soc à 65 % selon la procédure des médicaments d'exception (prescription en conformité avec la fiche d'information thérapeutique). Collect. |
|
| Prix ou tarif de responsabilité (HT) par UCD : | UCD 9311204 (seringue à 1000 UI/0,5 ml) : 7.371 euros. |
| UCD 9311227 (seringue à 2000 UI/1 ml) : 14.740 euros. | |
| UCD 9311233 (seringue à 3000 UI/0,3 ml) : 22.110 euros. | |
| UCD 9311256 (seringue à 4000 UI/0,4 ml) : 29.480 euros. | |
| UCD 9311262 (seringue à 5000 UI/0,5 ml) : 36.860 euros. | |
| UCD 9311279 (seringue à 6000 UI/0,6 ml) : 44.230 euros. | |
| UCD 9311285 (seringue à 8000 UI/0,8 ml) : 58.970 euros. | |
| UCD 9311210 (seringue à 10 000 UI/1 ml) : 73.710 euros. | |
| UCD 9343612 (seringue à 20 000 UI/0,5 ml) : 147.420 euros. | |
| UCD 9343629 (seringue à 30 000 UI/0,75 ml) : 221.130 euros. | |
| UCD 9343635 (seringue à 40 000 UI/1 ml) : 294.840 euros. | |
| Inscrit sur la liste des spécialités prises en charge en sus des GHS. | |
SANDOZ
49, av Georges-Pompidou
92593 Levallois-Perret cdx
Tél : 01 49 64 48 00
Info médic, pharmacovigilance et service client :
Tél (n° Vert) : 08 00 45 57 99
Site web : http://www.sandoz.fr
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale
