MACUGEN ®
pégaptanib
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p seringue | |
| Pégaptanib (DCI) sodium | 1,65 mg* |
INDICATIONS |
POSOLOGIE ET MODE D'ADMINISTRATION |
Posologie :
Les antécédents médicaux du patient relatifs aux réactions d'hypersensibilité doivent être attentivement évalués avant de procéder à l'administration intravitréenne (cf Mises en garde et Précautions d'emploi).
Macugen 0,3 mg doit être administré toutes les six semaines (9 injections par an) par injection intravitréenne dans l'oeil atteint.
Après l'injection, des élévations transitoires de la pression intraoculaire ont été observées chez les patients traités par Macugen. Par conséquent, la perfusion de la tête du nerf optique ainsi que la pression intraoculaire doivent être surveillées. De plus, les risques d'hémorragie vitréenne et d'endophtalmie doivent être étroitement surveillés chez les patients dans les deux semaines suivant l'injection. Les patients doivent être informés que tout symptôme évocateur de ces conditions doit être signalé sans délai (cf Mises en garde et Précautions d'emploi).
Lors de la visite à 12 semaines, si un patient ne démontre pas de bénéfice thérapeutique (perte de moins de 15 lettres d'acuité visuelle) après 2 injections consécutives de Macugen, l'arrêt ou le report du traitement par Macugen doit être pris en considération.
- Populations spécifiques :
-
- Personnes âgées :
- Aucune précaution particulière n'est nécessaire.
-
- Insuffisance hépatique :
- Macugen n'a pas été étudié chez les patients présentant une insuffisance hépatique. Cependant, aucune précaution particulière n'est nécessaire pour cette population (cf Pharmacocinétique).
-
- Insuffisance rénale :
- Macugen n'a pas été étudié suffisamment chez les patients présentant une insuffisance rénale sévère, il n'est toutefois pas recommandé d'effectuer un ajustement de la dose chez les patients présentant une insuffisance rénale faible ou modérée (cf Pharmacocinétique).
-
- Sexe :
- Aucune précaution particulière n'est nécessaire.
-
- Population pédiatrique :
- La sécurité et l'efficacité de Macugen chez l'enfant de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration :
Pour usage intravitréen uniquement.
Macugen doit être contrôlé visuellement avant l'administration pour vérifier l'absence de particules et de changement de coloration (cf Modalités de manipulation et d'élimination).
La procédure d'injection doit être réalisée en conditions d'asepsie, incluant la désinfection chirurgicale des mains, le port de gants stériles, l'utilisation d'un champ stérile et d'un spéculum à paupières stérile (ou équivalent), et la possibilité d'effectuer une paracentèse stérile (si nécessaire). Une anesthésie appropriée et un antibactérien local à large spectre doivent être administrés avant l'injection.
CONTRE-INDICATIONS |
- Hypersensibilité connue à la substance active ou à l'un des excipients.
- Infection oculaire ou périoculaire active ou suspectée.
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
Après les injections de pégaptanib, des hémorragies intravitréennes peuvent survenir immédiatement (le jour de l'injection) ou de façon retardée (cf Posologie et Mode d'administration).
Les procédures d'injection intravitréenne sont associées à un risque d'endophtalmie ; dans les études cliniques portant sur Macugen, l'incidence d'endophtalmie était de 0,1 % par injection (cf Posologie et Mode d'administration).
Après commercialisation, des cas de réactions anaphylactiques/anaphylactoïdes, dont oedème de Quincke, ont été observés plusieurs heures après l'administration intravitréenne de pégaptanib. Dans ces cas, il n'a pas été établi de lien direct ni avec Macugen, ni avec l'un des divers traitements administrés dans le cadre de la procédure de préparation à l'injection, ou avec tout autre facteur.
INTERACTIONS |
Les deux premières études cliniques, menées sur des patients ayant reçu Macugen seul et en association à la thérapie photodynamique (PDT), n'ont pas mis en évidence de différence dans la pharmacocinétique plasmatique du pégaptanib.
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Le pégaptanib n'a pas été étudié chez les femmes enceintes. Les études chez l'animal sont insuffisantes, mais ont montré une toxicité de la reproduction à des taux d'exposition systémique élevés (cf Sécurité préclinique). Le risque potentiel dans l'espèce humaine n'est pas connu. L'exposition systémique au pégaptanib, attendue après une administration oculaire, est très faible. Néanmoins, Macugen ne doit être utilisé pendant la grossesse que si le bénéfice potentiel pour la mère justifie le risque potentiel pour le foetus.
Allaitement :
L'excrétion de Macugen dans le lait maternel n'a pas été établie. L'administration de Macugen est déconseillée pendant l'allaitement.
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
374 patients ont reçu un traitement continu par Macugen sur une période allant jusqu'à deux ans (128 patients à 0,3 mg, 126 patients à 1 mg et 120 patients à 3 mg). Les données de tolérance globales étaient concordantes avec les données de tolérance à un an et aucun nouveau signal n'est apparu. Parmi les 128 patients qui avaient été traités jusqu'à deux ans avec la dose recommandée de 0,3 mg (nombre total d'injections la deuxième année : 913, nombre moyen d'injections la deuxième année : 6,9), il n'a pas été observé d'augmentation significative de la fréquence des événements indésirables comparativement à la fréquence observée pendant la première année.
Les effets indésirables oculaires graves rapportés chez les patients traités par Macugen comprennent des endophtalmies (12 cas, 1 %), des hémorragies rétiniennes (3 cas, < 1 %), des hémorragies vitréennes (2 cas, < 1 %) et des décollements de rétine (4 cas, < 1 %).
Les données de tolérance décrites ci-dessous résument tous les effets indésirables liés à la procédure et au médicament chez les 295 patients du groupe traité à 0,3 mg. Les effets indésirables sont listés par classe de système d'organe et par fréquence (très fréquent : >= 1/10 ; fréquent : >= 1/100 et < 1/10 ; et peu fréquent : >= 1/1000 et < 1/100), fréquence indéterminée [ne peut être estimée d'après les données disponibles]).
Les données obtenues après la commercialisation sont inclues en italique.
Affections du système immunitaire :
- Fréquence indéterminée : Réaction anaphylactique*.
- Peu fréquent : cauchemar, dépression.
- Fréquent : céphalées.
- Très fréquent : inflammation de la chambre antérieure, douleur oculaire, augmentation de la pression intraoculaire, kératite ponctuée, corps flottants et opacité du corps vitré.
- Fréquent : sensations anormales dans l'oeil, cataracte, hémorragie conjonctivale, hyperhémie conjonctivale, oedème conjonctival, conjonctivite, dystrophie de la cornée, atteinte de l'épithélium cornéen, affection de l'épithélium cornéen, oedème cornéen, sécheresse oculaire, endophtalmie, écoulement oculaire, inflammation oculaire, irritation oculaire, prurit oculaire, rougeur oculaire, gonflement de l'oeil, oedème de la paupière, sécrétion lacrymale accrue, dégénérescence maculaire, mydriase, gêne oculaire, hypertension oculaire, hématome périorbital, photophobie, photopsie, hémorragie rétinienne, vision floue, baisse d'acuité visuelle, trouble visuel, décollement du corps vitré et affection du corps vitré.
- Peu fréquent : asthénopie, blépharite, conjonctivite allergique, dépôts cornéens, hémorragie oculaire, prurit des paupières, kératite, hémorragie du corps vitré, altération des réflexes pupillaires, abrasion de la cornée, exsudats rétiniens, ptose de la paupière, cicatrice rétinienne, chalazion, érosion cornéenne, baisse de la pression intraoculaire, réaction au site d'injection, vésicules au site d'injection, décollement de la rétine, affection de la cornée, occlusion de l'artère rétinienne, déchirure rétinienne, ectropion, trouble du mouvement oculaire, irritation de la paupière, hyphéma, affection de la pupille, affection de l'iris, ictère oculaire, uvéite antérieure, dépôt oculaire, iritis, excavation du nerf optique, déformation pupillaire, occlusion de la veine rétinienne et prolapsus du corps vitré.
- Peu fréquent : surdité, aggravation de la maladie de Ménière, vertiges.
- Peu fréquent : palpitations.
- Peu fréquent : hypertension, anévrisme aortique.
- Fréquent : rhinorrhée.
- Peu fréquent : rhinopharyngite.
- Peu fréquent : vomissement, dyspepsie.
- Peu fréquent : dermatite de contact, eczéma, modification de la couleur des cheveux, rash, prurit, sueurs nocturnes.
- Fréquence indéterminée : Angio-oedème*.
- Peu fréquent : dorsalgie.
- Peu fréquent : fatigue, frissons, sensibilité douloureuse, douleur thoracique, syndrome grippal.
- Peu fréquent : élévation de l'activité des gamma-glutamyl transférases.
- Peu fréquent : abrasion.
* Après commercialisation : des cas de réactions anaphylactiques/anaphylactoïdes, dont oedème de Quincke, ont été rapportés plusieurs heures après l'administration intravitréenne de pégaptanib ainsi que divers médicaments administrés dans le cadre de la procédure de préparation à l'injection (cf Posologie et Mode d'administration, Mises en garde et Précautions d'emploi).
SURDOSAGE |
PHARMACODYNAMIE |
Classe pharmacothérapeutique : Produits ophtalmiques ; agents des troubles vasculaires oculaires (code ATC : S01LA03).
- Mécanisme d'action :
- Le pégaptanib est un oligonucléotide modifié pégylé qui se lie avec une haute spécificité et affinité au facteur de croissance vasculaire endothélial extracellulaire (VEGF165) en inhibant son activité. Le VEGF est une protéine sécrétée qui induit une angiogenèse, une perméabilité vasculaire et une inflammation, tous ces facteurs étant considérés comme contribuant à la progression de la forme néovasculaire (humide) de la DMLA.
- Propriétés pharmacodynamiques :
- Le VEGF165 est l'isoforme du VEGF impliquée préférentiellement dans la néovascularisation oculaire pathologique. Chez l'animal, l'inhibition sélective par le pégaptanib s'est révélée aussi efficace pour supprimer la néovascularisation pathologique que l'inhibition totale du VEGF, mais le pégaptanib a épargné le système vasculaire normal, contrairement à l'inhibition totale du VEGF. Des réductions de l'augmentation de la taille totale moyenne de la lésion, de la taille de la néovascularisation choroïdienne (NVC) et de la taille de la diffusion de la fluorescéine ont été observées chez les patients atteints de DMLA traités avec Macugen.
- Efficacité clinique et sécurité :
- Le pégaptanib a été étudié dans deux études identiques (EOP1003, EOP1004) randomisées, en double aveugle, contrôlées, menées sur des patients atteints de DMLA néovasculaire. Un total de 1190 patients d'un âge médian de 77 ans ont été traités (892 par Macugen, 298 par injection simulée [contrôle]). Au sein de tous les groupes de patients traités, les patients ont reçu en moyenne 8,4 à 8,6 injections sur un total de 9 injections possibles lors de la première année.
- Les patients ont été randomisés pour recevoir soit une injection intravitréenne simulée soit une injection intravitréenne de pégaptanib 0,3 mg, 1 mg ou 3 mg, toutes les 6 semaines pendant 48 semaines. Le traitement photodynamique par la vertéporfine (PDT) a été administré à la libre appréciation des investigateurs chez les patients présentant des lésions à prédominance visible.
- Les deux essais ont inclus des patients présentant tous les sous-types de lésions néovasculaires de la DMLA (25 % à prédominance visible, 39 % occulte sans néovaisseaux visibles et 36 % à peine visible), la taille des lésions allant jusqu'à des surfaces de disque de 12 pour lesquelles jusqu'à 50 % pouvaient être compliquées d'une hémorragie sous-rétinienne et/ou jusqu'à 25 % de cicatrice fibrotique ou de lésion atrophique. Les patients ont pu avoir une PDT préalable. L'acuité visuelle initiale de l'oeil étudié était comprise entre 20/40 et 20/320.
- Après un an, le pégaptanib 0,3 mg a montré un bénéfice thérapeutique statistiquement significatif pour le critère d'efficacité principal, la proportion de patients perdant moins de 15 lettres d'acuité visuelle (analyse groupée prédéfinie, pégaptanib 0,3 mg 70 % versus injection simulée 55 %, p = 0,0001 ; EOP1003 pégaptanib 0,3 mg 73 % versus injection simulée 59 %, p = 0,0105 ; EOP1004 pégaptanib 0,3 mg 67 % versus injection simulée 52 %, p = 0,0031).
-
Variation moyenne de l'acuité visuelle au cours du temps ; année 1 ; en ITT (intention de traiter) ; LOCF-dernière observation reportée : 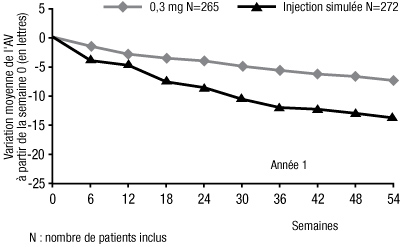
- Le pégaptanib 0,3 mg a montré un bénéfice thérapeutique quels que soient le sous-type de lésion, la taille des lésions, l'acuité visuelle, mais aussi quels que soient l'âge, le sexe, la pigmentation de l'iris au début de l'essai, et qu'il y ait eu une utilisation de la PDT avant l'essai et/ou au début de l'essai.
- A la fin de la première année (semaine 54), 1053 patients ont été randomisés à nouveau, soit pour continuer, soit pour arrêter le traitement jusqu'à la semaine 102.
- En moyenne, le bénéfice du traitement s'est maintenu à la semaine 102 avec une préservation persistante de l'acuité visuelle chez les patients rerandomisés pour la poursuite du traitement par le pégaptanib. Les patients rerandomisés pour l'arrêt du traitement par le pégaptanib après 1 an ont présenté une perte d'acuité visuelle lors de la deuxième année.
-
Résumé des variations moyennes de l'acuité visuelle à partir de la valeur initiale jusqu'aux semaines 6, 12, 54 et 102 (LOCF - dernière observation reportée) : EOP1003 EOP1004 0,3-0,3 0,3-arrêt Injection simulée-injection simulée/injection simulée + arrêt 0,3-0,3 0,3-arrêt Injection simulée-injection simulée/injection simulée + arrêt N 67 66 54 66 66 53 Variation moyenne de l'AV
Semaine 6- 1,9 - 0,0 - 4,4 - 1,9 - 2,0 - 3,4 Variation moyenne de l'AV
Semaine 12- 4,3 - 2,0 - 4,8 - 2,8 - 2,2 - 4,7 Variation moyenne de l'AV
Semaine 54- 9,6 - 4,3 - 11,7 - 8,0 - 7,6 - 15,6 Variation moyenne de l'AV
Semaine 102- 10,8 - 9,7 - 13,1 - 8,0 - 12,7 - 21,1 - Sur la période des deux ans, les données montrent que le traitement par Macugen doit être initié aussi précocement que possible. En cas de maladie avancée, l'initiation et la poursuite du traitement par Macugen doivent prendre en compte les risques pour une vision utile à l'oeil.
- L'administration de Macugen simultanément dans les deux yeux n'a pas été étudiée.
- La tolérance et l'efficacité de Macugen au-delà de deux ans n'ont pas été démontrées.
- L'Agence européenne du médicament a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec Macugen dans tous les sous-groupes de la population pédiatrique dans la dégénérescence maculaire liée à l'âge (cf Posologie et Mode d'administration pour les informations concernant l'usage pédiatrique).
PHARMACOCINÉTIQUE |
- Absorption :
- Chez l'animal, après une administration intravitréenne, le pégaptanib est lentement absorbé dans la circulation systémique à partir de l'oeil. Le taux d'absorption à partir de l'oeil constitue le facteur limitant de la disponibilité du pégaptanib chez l'animal, et probablement chez l'homme.
- Chez l'homme, la demi-vie plasmatique apparente moyenne ± écart type du pégaptanib après une dose monoculaire de 3 mg (soit 10 fois la dose recommandée) est de 10 jours ± 4. Chez l'homme, après une dose monoculaire de 3 mg, une concentration plasmatique maximale moyenne d'environ 80 ng/ml est observée dans les 1 à 4 jours. L'aire moyenne sous la courbe concentration plasmatique/temps (ASC) est d'environ 25 µg × h/ml à cette dose. Le pégaptanib ne s'accumule pas dans le plasma lorsqu'il est administré par injection intravitréenne toutes les 6 semaines. Aux doses inférieures à 0,5 mg/oeil, les concentrations plasmatiques du pégaptanib ne devraient pas être supérieures à 10 ng/ml.
- La biodisponibilité absolue du pégaptanib après une administration intravitréenne n'a pas été évaluée chez l'homme, mais elle est d'environ 70 à 100 % chez le lapin, le chien et le singe.
- Chez les animaux ayant reçu dans les deux yeux des doses du pégaptanib allant jusqu'à 0,5 mg/oeil, les concentrations plasmatiques étaient 0,03 % à 0,15 % de celles observées dans l'humeur vitrée.
- Distribution, biotransformation, excrétion :
- Chez la souris, le rat, le lapin, le chien et le singe, le pégaptanib est distribué principalement dans le volume plasmatique et il est peu distribué dans les tissus périphériques après une administration intraveineuse. Chez le lapin et vingt-quatre heures après une administration intravitréenne dans les deux yeux d'une dose radiomarquée de pégaptanib, la radioactivité était essentiellement distribuée dans l'humeur vitrée, la rétine et l'humeur aqueuse. Après des administrations intravitréennes et intraveineuses de pégaptanib radiomarqué à des lapins, les concentrations de radioactivité les plus élevées (à l'exception de l'oeil ayant reçu la dose par voie intravitréenne) ont été observées dans le rein. Chez le lapin, le composant nucléotidique 2'-fluorouridine est détecté dans le plasma et l'urine après l'administration intraveineuse ou intravitréenne d'une dose unique de pégaptanib radiomarqué. Le pégaptanib est métabolisé par des endo et des exonucléases. Chez le lapin, le pégaptanib est éliminé principalement dans l'urine sous forme de substance mère et de métabolites.
- Populations spécifiques :
- La pharmacocinétique du pégaptanib est identique chez les patients de sexe masculin ou féminin, et dans la tranche d'âge de 50 à 90 ans.
- Le pégaptanib sodium n'a pas été étudié suffisamment chez les patients présentant une clairance de la créatinine inférieure à 20 ml/min. Une diminution de la clairance de la créatinine en dessous de 20 ml/min peut être associée à une augmentation jusqu'à 2,3 fois de l'ASC du pégaptanib. Aucune précaution particulière n'est nécessaire pour les patients présentant une clairance de la créatinine supérieure à 20 ml/min traités avec la dose recommandée de 0,3 mg de pégaptanib sodium.
- La pharmacocinétique du pégaptanib n'a pas été étudiée chez les patients présentant une insuffisance hépatique. L'exposition systémique chez ce type de patients est supposée rester dans des limites bien tolérées, puisqu'une dose 10 fois plus élevée (3 mg/oeil) a été bien tolérée.
SÉCURITE PRÉCLINIQUE |
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, de toxicité en administration répétée et de génotoxicité n'ont pas révélé de risque particulier pour l'homme. Il n'existe pas d'études sur le potentiel carcinogène du pégaptanib.
Le pégaptanib n'a pas entraîné de toxicité maternelle ni démontré de tératogénicité ou de mortalité foetale chez la souris lors de l'administration intraveineuse de doses allant de 1 à 40 mg/kg/jour. Une diminution du poids corporel (5 %) et un retard minimal d'ossification des phalanges des pattes de devant ont été observés uniquement à des taux d'exposition basés sur une ASC de plus de 300 fois supérieure à celle attendue chez l'homme. Ces résultats sont donc considérés comme ayant une pertinence clinique limitée. Dans le groupe 40 mg/kg/jour, les concentrations du pégaptanib dans le liquide amniotique représentaient 0,05 % des taux plasmatiques maternels. Il n'y a pas d'études de toxicité de la reproduction chez le lapin.
Aucune donnée n'est disponible pour évaluer l'accouplement des mâles ou des femelles ou les indices de fertilité.
INCOMPATIBILITÉS |
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 3 ans.
A conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler.
MODALITÉS MANIPULATION/ÉLIMINATION |
Macugen est destiné à un usage unique exclusivement. Si la solution présente un aspect trouble, ou en présence de particules, ou de signes de dommages sur la seringue, ou bien si le clip de fixation en plastique est manquant ou n'est pas fixé à la seringue, Macugen ne doit pas être utilisé.
Avant l'administration, la seringue doit être retirée du clip de fixation en plastique et le capuchon enlevé. Une aiguille de 27 ou 30 G (12,7 mm) doit être fixée sur l'adaptateur luer-lock pour permettre l'administration du médicament.
La présence de bulles dans la seringue doit être vérifiée avec l'aiguille pointée vers le haut. S'il y a des bulles, la seringue doit être délicatement tapotée avec un doigt jusqu'à ce que les bulles remontent à la surface de la seringue. Puis, le piston doit être lentement poussé pour refouler les bulles à l'extérieur de la seringue. Le bouchon du piston ne doit pas être retiré.
La dernière nervure du bouchon du piston (la plus près de la tige du piston) ne doit pas être poussée au-delà du repère de la dose indiqué sur la seringue. Immédiatement avant l'administration, cette dernière nervure du bouchon doit être alignée avec le repère de la dose pour assurer la délivrance de la dose appropriée. A cette étape, tout le contenu de la seringue doit être injecté.
Macugen doit être conservé au réfrigérateur à une température comprise entre 2 °C et 8 °C. La solution à injecter doit atteindre la température ambiante avant d'être injectée. Macugen doit être jeté s'il reste à température ambiante plus de deux semaines. Afin de prévenir toute contamination, la seringue de Macugen ne doit pas être retirée de son étui avant que le patient n'ait été préparé pour l'injection.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| Médicament à prescription réservée aux spécialistes en ophtalmologie. | |
| AMM | EU/1/05/325/002 ; CIP 3400938260830 (2006, RCP rév 20.12.2010). |
| Prix : | 729.14 euros (1 seringue). |
|
Remb Séc soc à 100 % dans le traitement de la forme néovasculaire exsudative (humide) uniquement rétrofovéolaire de la dégénérescence maculaire liée à l'âge, selon la procédure des médicaments d'exception (prescription en conformité avec la fiche d'information thérapeutique). Le traitement par Macugen doit être exclusivement administré par injection intravitréenne et par des ophtalmologistes expérimentés dans ce type d'injection. Collect. |
|
PFIZER
23-25, av du Dr-Lannelongue. 75014 Paris
Tél : 01 58 07 30 00
Info médic : Tél : 01 58 07 34 40
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale
