TRISENOX®
arsenic
FORMES et PRÉSENTATIONS |
COMPOSITION |
| p ampoule | |
| Arsenic trioxyde | 10 mg |
INDICATIONS |
Le taux de réponse des autres sous-types de leucémie aiguë myéloblastique à Trisenox n'a pas été examiné.
POSOLOGIE ET MODE D'ADMINISTRATION |
Trisenox doit être administré sous la surveillance d'un médecin ayant l'expérience du traitement des leucémies aiguës ; d'autre part, les procédures inhérentes aux contrôles particuliers tels que décrits dans la rubrique Mises en garde/Précautions d'emploi doivent être suivies. La dose recommandée est identique pour les enfants, les adultes et les sujets âgés.
- Plan du traitement d'induction :
- Trisenox doit être administré par voie intraveineuse, à la dose fixe de 0,15 mg/kg/jour prise quotidiennement jusqu'à rémission médullaire (moins de 5 % de blastes présents dans la moelle osseuse riche en cellules, sans trace de cellules leucémiques). Si une rémission médullaire n'est pas intervenue après 50 jours, le traitement doit être interrompu.
- Plan du traitement de consolidation :
- Le traitement de consolidation doit commencer 3 à 4 semaines après la fin du traitement d'induction. Trisenox doit être administré par voie intraveineuse à la dose de 0,15 mg/kg/jour, 25 fois, réparties à raison de 5 jours par semaine, suivis par 2 jours d'interruption, et ce, pendant 5 semaines.
- Usage pédiatrique :
- Il existe peu de données cliniques sur l'usage de Trisenox en pédiatrie. Sur 7 patients âgés de moins de 18 ans (limites : 5 - 16 ans) recevant Trisenox à la dose conseillée de 0,15 mg/kg/jour, 5 patients ont obtenu une réponse complète. La tolérance et l'efficacité n'ont pas été étudiées chez les enfants de moins de 5 ans.
- Insuffisants hépatiques et/ou rénaux :
- Les données disponibles chez les patients souffrant d'insuffisance hépatique et d'insuffisance rénale, quel que soit leur degré, étant limitées, il est recommandé d'utiliser Trisenox avec prudence chez ces patients.
Mode d'administration :
Trisenox doit être administré en perfusion intraveineuse de 1 à 2 heures. La durée de la perfusion peut être portée à 4 heures en cas de réactions vasomotrices. Aucun cathéter veineux central n'est nécessaire. Les patients doivent être hospitalisés au début du traitement en raison des symptômes de la maladie et afin d'assurer une surveillance adéquate.
CONTRE-INDICATIONS |
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
- Syndrome d'activation des leucocytes (syndrome de différenciation LPA) :
- 25 % des patients atteints de LPA et traités par Trisenox ont présenté des symptômes analogues à ceux d'un syndrome appelé syndrome de l'acide rétinoïque-LPA (RA-APL) ou syndrome de différenciation LPA, caractérisé par une fièvre, une dyspnée, une prise de poids, des infiltrats pulmonaires et des épanchements pleuraux ou péricardiques, avec ou sans hyperleucocytose. Ce syndrome peut être fatal. Le traitement de ce syndrome n'a pas été défini de manière rigoureuse, mais l'administration de corticoïdes à hautes doses au premier signe de syndrome de différenciation LPA semble réduire les signes et symptômes. Dès les premiers signes évoquant ce syndrome (fièvre inexpliquée, dyspnée et/ou prise de poids, signes anormaux à l'auscultation thoracique ou anomalies radiographiques), une corticothérapie à hautes doses (dexaméthasone 10 mg par voie intraveineuse, deux fois par jour) doit être immédiatement instituée, quelle que soit la numération leucocytaire, et poursuivie pendant 3 jours ou plus jusqu'à ce que les signes et symptômes se soient atténués. Dans la majorité des cas, il n'est pas nécessaire d'arrêter l'administration de Trisenox pendant le traitement du syndrome de différenciation LPA. Il est recommandé que la chimiothérapie ne soit pas ajoutée à la corticothérapie, car il n'existe aucune expérience précédente d'administration conjointe de corticoïdes et d'une chimiothérapie durant le traitement du syndrome d'activation des leucocytes dû à Trisenox. L'expérience de post-commercialisation sous-tend qu'un syndrome similaire peut avoir lieu chez les patients atteints d'autres types de malignité. Les modes de traitement et de contrôle inhérents à ces patients doivent être tels que décrits ci-dessus.
- Anomalies de l'électrocardiogramme (ECG) :
- Le trioxyde d'arsenic peut occasionner une prolongation de l'intervalle QT et un bloc auriculoventriculaire complet. La prolongation de l'intervalle QT peut aboutir à une arythmie ventriculaire de type torsade de pointes, qui peut être fatale. Tout traitement antérieur à base d'anthracyclines peut accroître le risque de prolongation de l'intervalle QT. Le risque de torsade de pointes est lié aux facteurs suivants : degré de prolongation de l'intervalle QT, administration concomitante de produits prolongeant l'intervalle QT (tels que les antiarythmiques de classe la et III (ex : quinidine, amiodarone, sotalol, dofétilide), les antipsychotiques (ex : thioridazine), les antidépresseurs (ex : amitriptyline), certains macrolides (ex : érythromycine), certains antihistaminiques (ex : terfénadine et astémizole), certains antibiotiques de la famille des quinolones (ex : sparfloxacine) et autres médicaments connus pour prolonger l'intervalle QT (ex : cisapride), antécédents de torsade de pointes, prolongation préexistante de l'intervalle QT, insuffisance cardiaque congestive, administration de diurétiques éliminant le potassium, amphotéricine B ou autre affection entraînant une hypokaliémie ou une hypomagnésémie. Lors des essais cliniques, 40 % des patients traités par Trisenox ont présenté au moins une prolongation de l'intervalle QT corrigé (QTc) supérieure à 500 msec. Une prolongation de l'intervalle QTc a été observée 1 à 5 semaines après la perfusion de Trisenox, avec retour à la valeur initiale au terme de la 8e semaine suivant la perfusion de Trisenox. Une patiente (recevant plusieurs médicaments concomitants, dont l'amphotéricine B) a présenté un phénomène de torsade de pointes asymptomatique pendant le traitement d'induction d'une rechute de LPA par le trioxyde d'arsenic.
- Recommandations de contrôle de l'ECG et du profil électrolytique :
- Avant de commencer un traitement par Trisenox, un ECG à 12 dérivations sera pratiqué, ainsi qu'un dosage sérique des électrolytes (potassium, calcium et magnésium) et de la créatinine. Les anomalies électrolytiques préexistantes seront corrigées et, si possible, les traitements connus pour prolonger l'intervalle QT seront interrompus. Les patients présentant des facteurs de risque de prolongation de QTc ou des facteurs de risque de torsade de pointes devront faire l'objet d'une surveillance cardiaque continue (ECG). Pour QTc supérieur à 500 msec, des mesures correctives doivent être prises et QTc réévalué par des ECG en série avant d'envisager l'utilisation de Trisenox. Pendant le traitement par Trisenox, on veillera à maintenir constamment la kaliémie à plus de 4 mEq/l et la magnésémie à plus de 1,8 mg/dl. Les patients dont l'intervalle QT atteint une valeur absolue > 500 msec doivent être réévalués et une action immédiate sera entreprise pour corriger les éventuels facteurs de risque concomitants, alors qu'il faudra également évaluer les rapports bénéfices/risques de la poursuite et de l'arrêt du traitement par Trisenox. En cas de syncope ou d'accélération ou irrégularités du rythme cardiaque, le patient devra être hospitalisé et surveillé en continu, un dosage sérique des électrolytes sera pratiqué et le traitement par Trisenox sera suspendu jusqu'à ce que l'intervalle QTc repasse sous 460 msec, que les anomalies électrolytiques soient corrigées et que la syncope et les irrégularités du rythme cardiaque disparaissent. Il n'existe aucune donnée concernant l'effet de Trisenox sur l'intervalle QTc durant la perfusion. Un ECG sera effectué deux fois par semaine, et plus fréquemment pour les patients cliniquement instables, pendant les phases d'induction et de consolidation.
- Modification de la posologie :
- Le traitement par Trisenox devra à tout moment être suspendu, ajusté ou abandonné avant le terme programmé du traitement, dès lors qu'une toxicité de grade 3 ou plus (National Cancer Institute, Common Toxicity Criteria, Version 2) sera observée et jugée comme pouvant être liée au traitement par Trisenox. Les patients présentant de telles réactions considérées comme liées à Trisenox ne pourront reprendre le traitement qu'après résolution de l'effet toxique ou après retour à l'état initial de l'anomalie ayant provoqué l'interruption. Dans ce cas, le traitement devra reprendre à 50 % de la dose quotidienne précédente. Si l'effet toxique ne se reproduit pas dans les 3 jours suivant la reprise du traitement à la dose réduite, la dose quotidienne pourra repasser à 100 % de la dose originale. Le traitement sera abandonné chez les patients présentant une récurrence de la toxicité.
- Examens biologiques :
- Le profil électrolytique, la glycémie, ainsi que les bilans hématologique, hépatique, rénal et de coagulation du patient doivent être surveillés au moins deux fois par semaine, et plus fréquemment pour les patients cliniquement instables, pendant la phase d'induction, et au moins une fois par semaine pendant la phase de consolidation.
- Insuffisance rénale :
- Du fait des données limitées chez les patients présentant une insuffisance rénale, quelle qu'en soit la sévérité, il est recommandé d'utiliser Trisenox avec prudence chez les patients ayant une insuffisance rénale. L'expérience chez les patients ayant une insuffisance rénale sévère est insuffisante pour déterminer si une adaptation de la dose est requise.
- L'utilisation de Trisenox chez les patients dialysés n'a pas été étudiée.
- Insuffisance hépatique :
- Du fait des données limitées dans tous les groupes de patients présentant une insuffisance hépatique, il est recommandé d'utiliser Trisenox avec prudence chez les patients ayant une insuffisance hépatique.
- L'expérience chez les patients ayant une insuffisance hépatique sévère est insuffisante pour déterminer si une adaptation de la dose est requise.
- Patients âgés :
- Il existe peu de données cliniques sur l'usage de Trisenox au sein de la population âgée. Une prudence particulière est requise chez ces patients.
- Hyperleucocytose :
- Chez certains patients, le traitement par Trisenox a été associé à l'apparition d'une hyperleucocytose (>= 10 × 103/µl). Il n'est apparu aucune corrélation entre le nombre de globules blancs (GB) à la visite de référence et l'apparition d'une hyperleucocytose, pas plus qu'entre le nombre de GB à la visite de référence et le nombre maximal de GB. L'hyperleucocytose n'a jamais fait l'objet d'une chimiothérapie supplémentaire et a cédé spontanément à la poursuite du traitement par Trisenox. Le nombre des GB pendant le traitement de consolidation était inférieur par rapport à la période du traitement d'induction et était toujours inférieur à 10 × 103/µl, sauf chez un patient présentant un nombre de GB de 22 × 103/µl pendant le traitement de consolidation. Vingt patients (50 %) ont présenté une hyperleucocytose ; cependant, chez tous ces patients, le nombre de GB était en baisse ou avait retrouvé une valeur normale au moment de la rémission médullaire et aucune chimiothérapie cytotoxique ou leucophérèse n'a été nécessaire.
INTERACTIONS |
Il n'existe aucune étude formelle des interactions pharmacocinétiques entre Trisenox et les autres produits thérapeutiques. Une prolongation de l'intervalle QT/QTc est attendue sous traitement par Trisenox, et des cas de torsade de pointes et de bloc auriculoventriculaire complet ont été décrits. Les patients qui reçoivent ou ont reçu des composés connus pour provoquer une hypokaliémie ou une hypomagnésémie, comme les diurétiques ou l'amphotéricine B, peuvent avoir un risque supérieur de torsade de pointes. La prudence s'impose lorsque Trisenox est coadministré avec des médicaments connus pour prolonger l'intervalle QT/QTc, comme les macrolides, l'antipsychotique thioridazine ou les composés connus pour provoquer une hypokaliémie ou une hypomagnésémie. Des informations complémentaires sur les agents thérapeutiques prolongeant l'intervalle QT sont mentionnées dans la rubrique Mises en garde/Précautions d'emploi. L'influence de Trisenox sur l'efficacité des autres antileucémiques est inconnue.
GROSSESSE et ALLAITEMENT |
L'arsenic est excrété dans le lait maternel. En raison du risque de réactions indésirables graves causées par Trisenox chez le nourrisson allaité, l'allaitement doit être interrompu avant et pendant toute la durée d'administration du produit.
CONDUITE et UTILISATION DE MACHINES |
EFFETS INDÉSIRABLES |
Les réactions indésirables sérieuses étaient fréquentes (1 - 10 %) et attendues dans cette population. Les réactions indésirables sérieuses imputées à Trisenox étaient un syndrome de différenciation LPA (3), une hyperleucocytose (3), une prolongation de l'intervalle QT (4, dont 1 avec torsade de pointes), une fibrillation/un flutter auriculaire (1), une hyperglycémie (2) et différentes réactions indésirables sérieuses à type d'hémorragies, d'infections, de douleur, de diarrhée et de nausées.
En général, les événements indésirables survenant sous traitement tendaient à diminuer avec le temps, peut-être suite à l'amélioration de la maladie traitée. Les patients avaient tendance à mieux tolérer le traitement de consolidation et d'entretien que le traitement d'induction. Cela provient probablement de l'effet confondant créé autour des événements indésirables par le processus pathologique mal contrôlé, au début du traitement, ainsi que par les nombreux traitements concomitants indispensables pour maîtriser les symptômes et la morbidité.
Le tableau ci-dessous énumère les réactions médicamenteuses indésirables de grade 3 et 4 rapportées chez les 107 patients traités par Trisenox dans le cadre d'essais cliniques (la fréquence a été définie comme suit : fréquent >= 1/100 à < 1/10, peu fréquent >= 1/1000 à < 1/100).
Dans chaque groupement par fréquence, les effets indésirables sont présentés par ordre de gravité décroissant.
| Classe de système d'organes | Fréquent | Peu fréquent |
| Troubles sanguins et du système lymphatique | Neutropénie Thrombocytopénie | Neutropénie fébrile Hyperleucocytose Leucopénie |
| Troubles métaboliques et nutritionnels | Hyperglycémie Hypokaliémie | Hypermagnésémie Hypernatrémie Acidocétose |
| Troubles du système nerveux | Paresthésie | |
| Troubles cardiaques | Épanchement péricardique Tachycardie |
|
| Troubles vasculaires | Vascularite | |
| Troubles respiratoires, thoraciques et médiastinaux | Douleur pleurétique Dyspnée | Hémorragie des alvéoles pulmonaires Épanchement pleural Hypoxie |
| Troubles gastro-intestinaux | Diarrhée | |
| Troubles cutanés et des tissus sous-cutanés | Prurit Érythème |
|
| Troubles musculosquelettiques et des tissus conjonctifs | Douleur osseuse Arthralgie | Myalgie |
| Troubles généraux et affections liées au site d'administration | Pyrexie Fatigue | Douleur thoracique Douleur |
| Investigations | ECG : prolongation de l'intervalle QT Augmentation de l'ALT Augmentation de l'aspartate aminotransférase | Hyperbilirubinémie Hypomagnésémie |
Dans les études majeures, la mortalité par coagulation intravasculaire disséminée (CIVD) était très fréquente (> 10%), ce qui est conforme aux taux de mortalité précoce rapportés dans la littérature.
Le trioxyde d'arsenic est susceptible d'entraîner une prolongation de l'intervalle QT (cf Mises en garde/Précautions d'emploi), laquelle peut conduire à une arythmie ventriculaire du type torsade de pointes, qui peut être fatale. Le risque de torsade de pointes est lié à différents facteurs : degré de prolongation de l'intervalle QT, administration concomitante de médicaments prolongeant l'intervalle QT, antécédents de torsade de pointes, prolongation préexistante de l'intervalle QT, insuffisance cardiaque congestive, administration de diurétiques d'élimination potassique ou autres pathologies donnant une hypokaliémie ou une hypomagnésémie. Une patiente (recevant plusieurs médicaments concomitants, dont l'amphotéricine B) a présenté un phénomène de torsade de pointes asymptomatique pendant le traitement d'induction d'une rechute de LPA par le trioxyde d'arsenic. Elle est passée au traitement de consolidation sans autre manifestation de prolongation de l'intervalle QT.
La neuropathie périphérique, caractérisée par des paresthésies/dysesthésies, est un effet courant et bien connu de l'arsenic présent dans l'environnement. Seuls 2 patients ont arrêté précocement le traitement en raison de cet événement indésirable et un a continué à recevoir Trisenox dans le cadre d'un protocole ultérieur. 44 % des patients ont présenté des symptômes pouvant être associés à une neuropathie, la plupart ont été légers à modérés et ont régressé après l'arrêt du traitement par Trisenox.
Les événements indésirables graves suivants ont été identifiés lors de l'utilisation ayant suivi l'approbation de Trisenox et ont été inclus en prenant en considération la fréquence observée, la gravité et la relation causale possible avec Trisenox. Ils sont présentés ci-dessous par classe de système d'organes et par fréquence (les fréquences ont été définies comme suit : peu fréquent [>= 1/1000 à < 1/100], fréquence indéterminée [ne peut être estimée sur la base des données disponibles]).
| Classe de système d'organes | Peu fréquent | Fréquence indéterminée |
| Infection et infestations | Septicémie Pneumonie Herpès zoster | |
| Troubles du système sanguin et du système lymphatique | Anémie | Pancytopénie |
| Troubles nutritionnels et du métabolisme | Déshydratation Rétention hydrique | |
| Troubles psychiatriques | État confusionnel | |
| Troubles du système nerveux | Convulsions Vertiges | |
| Troubles de la vision | Vision floue | |
| Troubles cardiaques | Arrêt cardiaque Tachycardie ventriculaire Extrasystoles ventriculaires | |
| Troubles vasculaires | Hypotension | |
| Troubles respiratoires, thoraciques et médiastinaux | Pneumopathie | Syndrome de différenciation |
| Troubles gastro-intestinaux | Vomissements Douleurs abdominales | |
| Troubles cutanés et sous-cutanés | OEdème facial Rougeurs | |
| Troubles rénaux et urinaires | Insuffisance rénale | |
| Troubles d'ordre général et au niveau des sites d'administration | OEdème Frissons | |
| Investigations | Augmentation de la créatinine sanguine Prise de poids |
SURDOSAGE |
PHARMACODYNAMIE |
Classe pharmacothérapeutique : autres anticancéreux (code ATC : L01XX27).
- Mécanisme d'action :
- Le mécanisme d'action de Trisenox n'est pas complètement élucidé. Le trioxyde d'arsenic induit in vitro des altérations morphologiques et des fragmentations de l'acide désoxyribonucléique (ADN) caractéristiques de l'apoptose des cellules NB4 humaines de la leucémie promyélocytaire. Le trioxyde d'arsenic provoque également la lésion ou la dégradation de la protéine de fusion PML/RAR-alpha.
- Essais cliniques :
- Trisenox a été étudié chez 52 patients atteints de LPA, précédemment traités par une anthracycline et un rétinoïde, dans deux essais ouverts non comparatifs, à un seul groupe. L'un était une étude monocentrique (n = 12) et l'autre une étude multicentrique effectuée dans 9 centres (n = 40). Les patients de la première étude ont reçu une dose médiane de Trisenox de 0,16 mg/kg/jour (limites : 0,06 à 0,20 mg/kg/jour) et ceux de l'étude multicentrique une dose fixe de 0,15 mg/kg/jour. Trisenox a été administré en perfusions intraveineuses de 1 à 2 heures jusqu'à disparition complète des cellules leucémiques de la moelle osseuse, pendant 60 jours au maximum. Les patients obtenant une rémission complète ont reçu un traitement de consolidation par Trisenox consistant en 25 doses supplémentaires sur une période de 5 semaines. Le traitement de consolidation a commencé 6 semaines (limites : 3 à 8 semaines) après le traitement d'induction dans l'étude monocentrique et 4 semaines (limites : 3 à 6 semaines) après le traitement d'induction dans l'étude multicentrique. Par définition, la rémission complète (RC) était caractérisée par l'absence de cellules leucémiques visibles dans la moelle osseuse et par la reconstitution d'une formule leucocytaire et plaquettaire normale dans le sang périphérique.
- Les patients de l'étude monocentrique avaient rechuté après 1 à 6 traitements antérieurs et 2 patients avaient rechuté après une transplantation de cellules souches. Les patients de l'étude multicentrique avaient rechuté après 1 à 4 traitements antérieurs et 5 patients avaient rechuté après une transplantation de cellules souches. L'âge médian des patients était de 33 ans (limites : 9 à 75 ans) dans l'étude monocentrique et de 40 ans (limites : 5 à 73 ans) dans l'étude multicentrique.
- Les résultats sont résumés dans le tableau ci-dessous.
-
Étude monocentrique
N = 12Étude multicentrique
N = 40Dose de Trisenox, mg/kg/jour (médiane, limites) 0,16 (0,06-0,20) 0,15 Rémission complète 11 (92 %) 34 (85 %) Délai de rémission médullaire (médiane) 32 jours 35 jours Délai de RC (médiane) 54 jours 59 jours Survie à 18 mois 67 % 66 % - L'étude monocentrique comprenait 2 enfants (< 18 ans) et tous deux ont obtenu une rémission complète (RC). L'essai multicentrique comprenait 5 enfants (< 18 ans), dont 3 ont obtenu une RC. Aucun enfant de moins de 5 ans n'a été traité.
Dans le suivi après le traitement de consolidation, 7 patients de l'étude monocentrique et 18 patients de l'étude multicentrique ont reçu un traitement d'entretien par Trisenox. 3 patients de l'étude monocentrique et 15 patients de l'étude multicentrique ont reçu une transplantation de cellules souches après avoir terminé le traitement par Trisenox. La durée médiane de la RC, selon la méthode de Kaplan-Meier, est de 14 mois pour l'étude monocentrique (elle n'a pas été atteinte pour l'étude multicentrique). A la dernière visite de contrôle, 6 patients sur 12 étaient vivants dans l'étude monocentrique, avec un suivi médian de 28 mois (limites : 25 à 29 mois). Dans l'étude multicentrique, 27 patients sur 40 étaient vivants, avec un suivi médian de 16 mois (limites : 9 à 25 mois).
| Les estimations de Kaplan-Meier de la survie à 18 mois pour chaque étude sont présentées ci-dessous. |
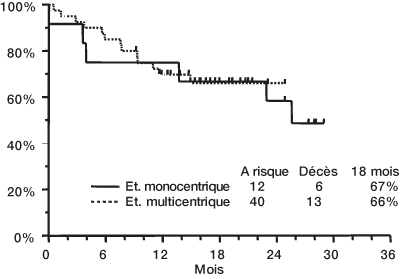 |
Le tableau suivant présente la confirmation cytogénétique de la conversion à un génotype normal et la détection par RT-PCR (Reverse Transcriptase-Polymerase Chain Reaction) de la conversion du facteur PML/RAR-alpha à la normale.
Analyse cytogénétique après traitement par Trisenox :
| Étude pilote monocentrique N avec RC = 11 | Étude multicentrique N avec RC = 34 |
|
| Cytogénétique classique [t(15;17)] | ||
| Absente | 8 (73 %) | 31 (91 %) |
| Présente | 1 (9 %) | 0 % |
| Non évaluable | 2 (18 %) | 3 (9 %) |
| RT-PCR pour PML/RARalpha | ||
| Négative | 8 (73 %) | 27 (79 %) |
| Positive | 3 (27 %) | 4 (12 %) |
| Non évaluable | 0 | 3 (9 %) |
Des réponses ont été observées dans toutes les tranches d'âge étudiées, allant de 6 à 75 ans. Les taux de réponses étaient similaires dans les deux sexes. Il n'existe aucune expérience de l'effet de Trisenox sur la variante de la LPA caractérisée par la présence des translocations chromosomiques t(11;17) et t(5;17).
PHARMACOCINÉTIQUE |
La forme inorganique, lyophilisée du trioxyde d'arsenic, mise en solution, forme immédiatement le produit d'hydrolyse : l'acide arsénieux (AsIII). AsIII est la forme pharmacologiquement active du trioxyde d'arsenic.
Dans l'intervalle de dose unique totale de 7 à 32 mg (administrée à 0,15 mg/kg), l'exposition systémique (AUC) apparaît linéaire. La décroissance à partir de la concentration du pic plasmatique d'AsIII intervient de manière biphasique et est caractérisée par une phase initiale de distribution rapide suivie par une phase terminale d'élimination lente. Après administration de 0,15 mg/kg à une fréquence journalière (n = 6) ou bihebdomadaire (n = 3), l'accumulation d'AsIII a été approximativement doublée par rapport à celle observée en administration unique. Cette accumulation était légèrement supérieure à ce qui était attendu sur la base des résultats en dose unique.
- Distribution :
- Le volume de distribution (Vd) d'AsIII est élevé (> 400 l) indiquant une distribution significative dans les tissus, avec une liaison négligeable aux protéines. Vd est aussi dépendant du poids, augmentant avec l'augmentation du poids corporel. L'arsenic total s'accumule principalement dans le foie, le rein et le coeur, et dans une moindre mesure, dans le poumon, les cheveux et les ongles.
- Métabolisme :
- Le métabolisme du trioxyde d'arsenic implique l'oxydation de l'acide arsénieux (AsIII), la forme active du trioxyde d'arsenic, en acide arsénique (AsV) ainsi que la méthylation oxydative en acide monométhylarsonique (MMAV) et en acide diméthylarsinique (DMAV) par des méthyltransférases, essentiellement dans le foie. Les métabolites pentavalents, MMAV et DMAV, sont lents à apparaître dans le plasma (environ 10-24 heures après la première administration de trioxyde d'arsenic), mais du fait de leur demi-vie longue, ils s'accumulent plus à des doses multiples que l'AsIII. L'étendue de l'accumulation de ces métabolites dépend du régime posologique. L'accumulation suivant une administration de dose multiple est approximativement de 1,4 à 8 fois supérieure à celle suivant une administration en dose unique. AsV est présent uniquement dans le plasma à des niveaux relativement faibles.
- Dans des études enzymatiques menées in vitro sur microsomes hépatiques humains, il a été démontré que le trioxyde d'arsenic ne possédait pas d'activité inhibitrice sur les substrats des enzymes principaux de cytochromes P450 tels que 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5, 4A9/11. Les médicaments substrats de ces enzymes P450 ne sont pas censés induire une interaction avec le Trisenox.
- Élimination :
- Environ 15 % de la dose administrée de Trisenox est excrétée dans les urines en AsIII inchangé. Les métabolites méthylés de l'AsIII (MMAV, DMAV) sont principalement excrétés dans les urines. La concentration plasmatique de l'AsIII décline de manière biphasique à partir de la concentration du pic plasmatique avec une demi-vie moyenne d'élimination terminale de 10 à 14 heures. La clairance totale de l'AsIII pour un intervalle de dose unique de 7-32 mg (administrée à 0,15 mg/kg) est de 49 l/h. La clairance n'est pas dépendante du poids du sujet ni de la dose administrée pour l'intervalle de dose étudié. Les demi-vies moyennes d'élimination terminale estimées des métabolites MMAV et DMAV sont respectivement de 32 heures et de 70 heures.
- Insuffisance rénale :
- La clairance plasmatique de l'AsIII n'a pas été altérée chez les patients présentant une insuffisance rénale faible (clairance de la créatinine de 50-80 ml/min) ou une insuffisance rénale modérée (clairance de la créatinine de 30-49 ml/min). La clairance plasmatique de l'AsIII chez les patients ayant une insuffisance rénale sévère (clairance de la créatinine inférieure à 30 ml/min) était 40 % inférieure à celle de patients ayant une fonction rénale normale (cf Mises en garde/Précautions d'emploi).
- L'exposition systématique au MMAV et au DMAV tend à être plus importante chez les patients ayant une insuffisance rénale ; la conséquence clinique en est inconnue mais aucune augmentation de la toxicité n'a été notée.
- Insuffisance hépatique :
- Des données pharmacocinétiques chez des patients ayant une insuffisance hépatique faible à modérée avec un carcinome hépatocellulaire indiquent que l'AsIII ou l'AsV ne s'accumulent pas lorsque les injections sont bihebdomadaires. Sur la base des AUC doses-normalisées (par mg de dose), l'altération de la fonction hépatique ne semble pas augmenter l'exposition systémique de l'AsIII, AsV, MMAV ou DMAV.
SÉCURITE PRÉCLINIQUE |
Des études limitées chez l'animal de toxicité sur la reproduction avec le trioxyde d'arsenic indiquent des propriétés embryotoxiques et tératogènes (anomalies du tube neural, anophtalmie et microphtalmie) en cas d'administration de 1 à 10 fois la dose clinique recommandée (mg/m2). Il n'a pas été conduit d'études de fertilité avec Trisenox. Les arsénieux induisent des aberrations chromosomiques et des transformations morphologiques dans des cellules de mammifères in vitro et in vivo. Il n'a pas été conduit d'études formelles de potentiel carcinogène avec le trioxyde d'arsenic. Cependant, le trioxyde d'arsenic et d'autres arsénieux inorganiques sont reconnus comme cancérogènes chez l'homme.
INCOMPATIBILITÉS |
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments, à l'exception de ceux mentionnés dans la rubrique Modalités de manipulation/Élimination.
MODALITÉS DE CONSERVATION |
- Durée de conservation :
- 4 ans.
Ne pas congeler.
Après dilution des solutions intraveineuses, Trisenox est chimiquement et physiquement stable pendant 24 heures, entre 15 °C et 30 °C, et 48 heures conservé au réfrigérateur (entre 2 °C et 8 °C). D'un point de vue microbiologique, le produit doit être utilisé immédiatement. S'il n'est pas utilisé immédiatement, les durées et conditions de conservation avant utilisation relèvent de la responsabilité de l'utilisateur et ne dépasseraient normalement pas 24 heures entre 2 °C et 8 °C, à moins que la dilution se soit déroulée dans des conditions d'asepsie contrôlées et validées.
MODALITÉS MANIPULATION/ÉLIMINATION |
- Préparation de Trisenox :
- Une technique aseptique doit être strictement observée durant la manipulation de Trisenox car il ne contient aucun conservateur.
- Trisenox doit être dilué dans 100 à 250 ml de sérum glucosé à 50 mg/ml (5 %) pour injections, ou de chlorure de sodium à 9 mg/ml (0,9 %) pour injections, immédiatement après ouverture de l'ampoule.
- Usage individuel.
- Toute fraction inutilisée de chaque ampoule doit être jetée en respectant les mesures de sécurité. Ne conserver aucune fraction inutilisée pour l'administrer ultérieurement.
- Trisenox ne doit pas être mélangé avec ou administré en même temps et dans la même sonde intraveineuse que d'autres médicaments.
- Trisenox doit être administré en perfusion intraveineuse lente de 1 à 2 heures. La durée de la perfusion peut être portée à 4 heures en cas de réactions vasomotrices. Il n'est pas nécessaire de mettre en place un cathéter veineux central.
- La solution diluée doit être limpide et incolore. L'absence de particules et de décoloration doit être contrôlée visuellement dans toute solution parentérale avant administration.
- Ne pas utiliser la préparation en cas de présence de particules étrangères.
- Procédure correcte d'élimination :
- Tout produit non utilisé, tout élément entrant en contact avec le produit ou tout déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| Médicament réservé à l'usage hospitalier. | |
| AMM | EU/1/02/204/001 ; CIP 3400956362059 (RCP rév 10.08.2010). |
| Collect. |
| Prix ou tarif de responsabilité (HT) par UCD : | UCD 9242911 (ampoule) : 393.52 euros. |
| Inscrit sur la liste des spécialités prises en charge en sus des GHS. | |
CEPHALON FRANCE
20, rue Charles-Martigny. 94700 Maisons-Alfort
Tél : 01 49 81 81 00
Info médic et scientifique :
Tél : 01 49 81 81 81
Liste Des Sections Les Plus Importantes :
- pathologies
- Medicaments
- Medicaments injectables
- Traitement D’Urgence
- Guide Infirmier Des Examens De Laboratoire
- Infirmiers En Urgences
- Fiche Technique Medical
- Techniques De Manipulations En Radiologie Medicale
- Bibliotheque_medicale
